

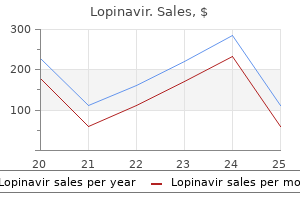
Lopinavir
| Contato
Página Inicial
Jessica Berrios, MD
- Department of Emergency Medicine
- Nassau University Medical Center
- East Meadow, New York
Because we have effective means of monitoring newborn hydration (such as a daily weight and measurements of serum electrolytes) symptoms mold exposure order generic lopinavir online, there is no reason to provide additional fluids routinely to infants who are receiving phototherapy medicine 75 purchase lopinavir now. They should symptoms your period is coming 250 mg lopinavir fast delivery, of course medications that cause tinnitus discount 250 mg lopinavir overnight delivery, be kept adequately hydrated medicine cabinets buy discount lopinavir 250 mg, not because phototherapy produces dehydration but because adequate urine output is important for effective phototherapy. The bilirubin isomer, lumirubin, is excreted in the bile and the urine, and lumirubin excretion appears to be an important element in the bilirubinlowering function of phototherapy (Ennever, 1990). Perhaps the most important clinical complication encountered during the use of phototherapy is that associated with the presence of direct hyperbilirubinemia or cholestatic jaundice. When infants with direct hyperbilirubinemia are exposed to phototherapy, they may develop a dark, grayish-brown discoloration of the skin, serum, and urine. The pathogenesis of this syndrome is unknown but is possibly related to the accumulation of porphyrins or other pigments in the plasma in the presence of cholestasis (Onishi et al, 1982; Rubaltelli et al, 1983, 1996). This rate has been demonstrated to be both safe and efficient (Forfar, 1958) and avoids deleterious hemodynamic changes (Aranda, 1977). Symptomatic hypocalcemia in early studies was reported in up to 5% of healthy infants, but supplemental calcium gluconate administration during the exchange transfusion has little effect on serum ionized calcium (Ellis, 1979; Maisels, 1974; Wieland, 1979), and too rapid infusion of calcium may cause bradyarrhythmias or cardiac arrest. If symptomatic hypocalcemia develops, temporary cessation of the procedure will allow recovery toward normal calcium levels as the citrate (which binds calcium) is metabolized by the liver. Postexchange studies should include bilirubin, hemoglobin, platelet count, ionized calcium, serum electrolytes, and serum glucose. The unintended consequences of exchange transfusion include cardiovascular, hematologic, gastrointestinal, biochemical, and infectious hazards, among others (Watchko, 2000c). Previously reported overall mortality rates associated with exchange transfusion ranged from 0. These rates, however, may not be generalizable to the current era if, as with most procedures, frequency of performance is an important determinant of risk and experience with exchange transfusion is decreasing (Newman e al, 1992). It is quite possible that the mortality (and morbidity) for this now infrequently performed procedure might be considerably higher than previously reported. On the other hand, none of the reports before 1986 included contemporary monitoring capabilities such as pulse oximetry. Jackson (1997) reported a 2% overall mortality rate (2 of 106) associated with exchange transfusions between 1980 and 1995 and a 12% risk of serious complications attributable to exchange transfusion in ill infants. Moreover, in infants classified as ill with medical problems in addition to hyperbilirubinemia, the incidence of exchange transfusion related complication leading to death was 8%. Symptomatic hypocalcemia, bleeding related to thrombocytopenia, catheter-related complications, and apnea-bradycardia requiring resuscitation were common serious morbidities observed in this study, suggesting that exchange transfusion should be performed by experienced individuals in a neonatal intensive care unit with continuous monitoring (including pulse oximetry) prepared to respond to these adverse events (Jackson, 1997). Finally, although the risk of blood transfusion is now very low, transfusion always carries some infection risk (Schreiber et al, 1996). Kaplan M, Hammerman C: Bilirubin and the genome: the hereditary basis of unconjugated neonatal hyperbilirubinemia, Curr Pharmacogenomics 3:21-42, 2005. Subcommittee on Hyperbilirubinemia: Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation, Pediatrics 114:297-316, 2004. Valaes T: Problems with prediction of neonatal hyperbilirubinemia, Pediatrics 108:175-177, 2001. Matthay Neonatal malignancies differ in incidence, clinical behavior, and heritable features from cancers seen in older children. Whereas acute leukemia is the most common malignancy in young children, the majority of neonatal tumors are solid tumors, many of which are detected prenatally during routine ultrasonography. Some childhood malignancies that carry excellent prognoses, such as acute lymphoblastic leukemia, are often fatal in neonates. In contrast, neuroblastoma, which responds poorly to treatment in older children, often spontaneously regresses in newborns. Among these are differences in drug metabolism in newborns, the sensitivity of rapidly growing normal tissues to chemotherapeutic agents and radiation, and the increased possibility of late effects including neurocognitive sequelae, impaired reproductive capacity, growth disturbances, and secondary malignancies. The epidemiology, etiology, and diagnosis of neonatal malignancy are reviewed here, followed by a discussion of commonly encountered malignancies. Incidence rates for the most common types of malignancy in infants are shown in Table 80-2. The mortality rates for infants with cancer exceed those for older children, even among similar histologic groups (Ries et al, 1999). Two notable exceptions are neuroblastoma, for which 5-year survival in newborns with disseminated disease is >90%, and infantile fibrosarcoma, for which cure rates often exceed those achieved in older children or adults. Although trend analyses suggest that the incidence of malignancy in the pediatric population may be increasing (Linabery and Ross, 2008), a number of factors affect incidence rates, including improvements in molecular methods of diagnosis, changes in population characteristics, screening fetal ultrasound practices, and case ascertainment by cancer registries (Spector and Linabery, 2009). The most common malignancy in infants is neuroblastoma, followed by leukemia, central nervous system tumors, retinoblastoma, and germ cell tumors (Linabery and Ross, 2008). Female and male infants have similar cancer incidence rates, but white infants have significantly higher rates than those reported in African American infants for all histologic types. An acquired or inherited abnormality of a cancerpredisposing gene that is critical during embryogenesis underlies many cases of neonatal cancer. Malignant transformation of normal cells results from the activation or suppression of these cancer-predisposing genes. The retinoblastoma gene at 13q is an example of a constitutional chromosomal abnormality that results in a high risk of malignancy. A number of well-defined hereditary conditions are associated with an increased incidence of specific neoplasms; these are listed in Table 80-3. A lack of family history should not dissuade the clinician from investigating these syndromes, because both spontaneous germline mutations and parental mosaicism occur. Infants <1 Year in Newborns, Infants, and Children Malignancy Leukemia Central nervous system tumors Neuroblastoma Lymphoma Renal tumors Sarcoma Hepatic tumors Teratoma Retinoblastoma Other Newborns <30 d (%) 13 3 54 0. These syndromes are typified by macroglossia, gigantism, and abdominal wall defects; patients may also have visceromegaly, flame nevus, neonatal hypoglycemia, microcephaly, and retardation (Scott et al, 2006). Also reported are rhabdomyosarcoma, neuroblastoma, ganglioneuroma, and adenomas and hamartomas. Transplacental Tumor Passage A rare cause of cancer in neonates and infants is the transplacental passage of tumor cells from the mother. Fewer than 20 cases of transplacentally transmitted cancer have been reported (Walker et al, 2002). Malignancies transmitted include leukemia, melanoma, lymphoma, hepatic carcinoma, and lung cancer. Transplacentally acquired neoplasm is usually apparent at birth or shortly thereafter, but diagnosis has been reported as late as age 8 months (Maruko et al, 2002). The frequency of malignancy in pregnant women is estimated at 1 per 1000 pregnancies (Greenlund et al, 2001; Maruko et al, 2004; Pavlidis, 2002). That transplacental transmission is so rare is attributed to the protective function of the placenta. Twin-to-Twin Transmission the risk of development of leukemia is increased in a monozygotic twin. If one monozygotic twin has leukemia, the co-twin has an approximately 25% chance of developing leukemia, usually within weeks or months of the diagnosis of the sibling. Growing evidence suggests that this increased incidence is likely due to in utero twin-to-twin transmission of a preleukemic clone rather than to the simultaneous development of a shared germline mutation facilitating the later development of leukemia (Greaves et al, 2003; Mahmoud et al, 1995). Environmental Factors Environmental factors are probably less important in the development of neonatal cancer compared with their role in the development of cancer in older children and adults. Exposure to ionizing radiation during pregnancy is known to increase the risk of a number of tumors, including acute leukemia, in exposed offspring. There appears to be a dose-response relationship between the dose of ionizing radiation received by the fetus in utero and the subsequent development of cancer in childhood, with doses on the order of 10 mGy sufficient to produce an increase in risk (Doll and Wakeford, 1997). Maternal exposure to drugs during pregnancy has been associated with the subsequent development of cancer in offspring. Maternal use of diethylstilbestrol has been strongly associated with the development of clear cell adenocarcinoma of the vagina and cervix in daughters born from those pregnancies (Herbst et al, 1971). A number of substances known to be teratogenic also may be carcinogenic to offspring. In utero exposure to phenytoin or other antiepileptic drugs can result in the fetal hydantoin syndrome; some infants with this syndrome have developed neuroblastoma (Ehrenbard and Chaganti, 1981). Excessive maternal alcohol consumption may be linked to an increased risk of developing cancer in the newborn period, particularly acute myeloid leukemia (Shu et al, 1996). The use of fertility drugs does not appear to increase the risk of cancer in the exposed offspring (Basatemur and Sutcliffe, 2008). Environmental exposures of the mother or father to hydrocarbons, dyes, and other chemicals and solvents may be related to the development of neonatal tumors, but there is only a weak association for most of the risk factors identified. The association of neoplasms with other environmental factors, such as maternal use of tobacco, has not been conclusively proven (Shu et al, 1996). Symptoms of malignancy in neonates can be nonspecific, such as irritability, poor feeding, failure to thrive, and fever. Table 80-4 lists clinical features associated with the more common malignancies found in the neonatal period. Laboratory and pathologic evaluations should be directed at making the diagnosis efficiently, sparing the newborn unnecessary procedures that can result in acute and chronic morbidity. Urine catecholamine excretion should be measured when neuroblastoma is being considered. Consultation with a pediatric oncologist should be obtained for help in making the initial diagnosis. Surgeons and pathologists should submit biopsy tissue for histologic examination, immunoperoxidase staining, flow cytometry, cytogenetic analysis, and tumor banking. In infants, the first clinical manifestations in more than half of cases result from the presence of metastatic disease rather than the primary tumor. However, despite the occurrence of widespread disease, neuroblastoma in newborn infants is almost always associated with biologically favorable features and carries a remarkably good prognosis. Neuroblastoma usually occurs sporadically, likely following random genetic mutations; environmental factors do not appear to play a significant role. These cases may result from a hereditary-predisposition locus now attributed to multiple different genetic aberrations. Chromosome 16p12-13 was identified as a likely predisposition locus, though no causal gene has been identified (Kushner et al, 2005; Maris et al, 2002). Familial neuroblastoma is inherited in an autosomal dominant mendelian fashion with incomplete penetrance. Neuroblastoma has also been seen in several patients with constitutional chromosomal rearrangements, including deletions overlapping putative tumor suppressor loci at chromosome bands 1p36 and 11q14-23. Clinical Manifestations Neuroblastoma may manifest as a tumor mass anywhere sympathetic neural tissue normally occurs. The clinical presentation can range from an asymptomatic newborn with an incidentally noted mass on prenatal ultrasound to a critically ill infant with massive hepatomegaly and respiratory distress. In the newborn, neuroblastoma most commonly manifests by enlargement of the liver alone, seen in 65% of cases, followed with subcutaneous metastases, seen in 32%. These percentages differ strikingly from those for older infants and children (Table 80-5). Metastases to lungs, bones, skull, and orbit are rare in the newborn, although clumps of tumor cells are often found in the bone marrow. The most common site for the primary tumor is within the abdomen, arising in the adrenal medulla or a sympathetic ganglion. The tumor may also arise in the posterior mediastinum with resultant bronchial obstruction or invasion of the neural foramina, with neurologic symptoms. Increasing dyspnea, cough, wheezing, or pulmonary infection may be the presenting sign or symptom. Metastatic lesions, especially of the skin and liver, are common presenting features in the neonatal period; in the newborn, the primary site often cannot be discovered. Massive liver involvement in newborns with disseminated neuroblastoma is responsible for the higher mortality rate in newborns compared with older infants (Nickerson et al, 2000). Subcutaneous skin nodules, which may be present at birth, are typically bluish in color. On palpation, the skin nodules may become erythematous for 2 to 3 minutes, followed by blanching of the lesion. The blanching is presumably due to vasoconstriction caused by release of catecholamines from the tumor cell and may be a diagnostic sign of subcutaneous neuroblastoma. Neuroblastoma arising from a paravertebral sympathetic ganglion has a tendency to grow into the intervertebral foramina, causing spinal cord compression and resultant paralysis. Careful periodic neurologic evaluation should be performed on a child with a neuroblastoma arising in this location, because the onset of cord compression may necessitate emergency intervention with chemotherapy, surgery, or irradiation. Unusual Presentations Intractable diarrhea can be the sole presenting manifestation of neuroblastoma. Secretion of vasoactive intestinal peptide by the tumor has been postulated to be the cause of the diarrhea, which resolves following surgical removal of the tumor (Bourdeaut et al, 2009). Removal of the tumor usually results in a decrease in neurologic signs and symptoms, but the use of steroids is frequently required for complete resolution. In general, the prognosis for survival of children with opsomyoclonus is excellent, although long-term neurologic deficits and learning delays are common and can be quite debilitating. Maternal symptoms such as sweating, pallor, headaches, palpitations, hypertension, and tingling in the feet and hands during the 8th and 9th months of pregnancy may be a sign of neuroblastoma in the fetus (Newton et al, 1985). The symptoms, which disappear postpartum, are likely caused by fetal catecholamines entering the maternal circulation. Newborns with neuroblastoma whose mothers have experienced these symptoms are typically diagnosed with neuroblastoma shortly after birth or during the first few months of life. Erythroblastosis with hepatosplenomegaly, severe jaundice, and an increase in nucleated red blood cells is occasionally seen in newborns with neuroblastoma. Congenital neuroblastoma with metastases to the liver and placenta can be clinically indistinguishable from hydrops fetalis. Catecholamine Secretion A hallmark of neuroblastoma cells is the ability to store and secrete catecholamines.
The increased contralateral testis volume symptoms 1974 purchase lopinavir 250 mg with mastercard, however silicium hair treatment purchase 250 mg lopinavir, does not preclude the need for exploration treatment 3rd degree burns lopinavir 250 mg order mastercard. Parents need to be reminded that in 80% of cases of inguinal exploration for an impalpable testis medicine jobs generic 250 mg lopinavir otc, the testis is located and a successful orchiopexy is performed medications a to z buy discount lopinavir on line. The child who presents at 6 months of age with an undescended testis high in the inguinal canal is also a candidate for surgical intervention, because the testis is unlikely to descend spontaneously. If the testis is near the scrotum, a repeat examination is prudent before pursuing surgical repair. Long-term issues associated with cryptorchidism include an increased risk of infertility and testicular malignancy. Fertility rates vary greatly depending on whether cryptorchidism is unilateral or bilateral. Furthermore, paternity was not affected by the age at the time of surgery (Lee et al, 1995). However, paternity rate decreased to 50% for patients with bilateral undescended testes who underwent orchiopexy in the first 3 years of life. As age at the time of bilateral orchiopexy increases, the paternity rates decline (Lee et al, 1997). A number of studies have demonstrated that cryptorchidism results in lower sperm counts despite surgical correction, and that the lowest counts are observed in patients with bilateral cryptorchidism. Testicular biopsy at the time of orchiopexy may offer a potential to identify those patients at the greatest risk of infertility 20 and 30 years later; a histologic scoring system allowing the prediction of a high-risk cohort of patients was recently validated with long-term follow-up semen analyses (Rusnack et al, 2003). The ability to predict low sperm counts using biopsy criteria also may allow for early hormonal treatment to enhance future fertility prospects in this select group of patients. A genetic contribution is suggested by the presence of a family history in 5% to 10% of cases (Chacko and Barthold, 2009). Antiandrogen treatment increased the likelihood of cryptorchidism in a rat model offering support for an environmental exposure hypothesis (Spencer et al, 1991). Deletion of the insulin 3 gene in mice results in cryptorchidism that, if untreated, results in infertility; however, microsurgical orchiopexies in this population of mice improved testicular histology and enhanced fertility (Nguyen et al, 2002). This work offers a scientific basis for orchiopexy, which remains one of the more commonly performed pediatric surgical procedures. In human studies of cryptorchidism, mutations in the insulin-3 gene are rare (Baker et al, 2002), suggesting that other pathways or mutations downstream of insulin-3 are responsible for this phenotype. The relative risk for testicular cancer in patients with an undescended testis increases approximately eightfold over that in the normal population, yet this observation is tempered by the rarity of testicular cancer. Thus, the overall lifetime risk of developing testicular cancer remains low for the individual patient, and in a large Scandinavian series, the incidence of malignancy was further diminished if surgical intervention was performed before puberty (Pettersson et al, 2007). Therefore, orchiopexy is recommended to place the testes in the proper scrotal position and to allow for monthly self-examination once the patient enters puberty. This event often occurs during the last trimester, and thus the indurated hemiscrotum observed in the delivery room is a late presentation of a terminal process. If the antenatal torsion occurs early in gestation, all inflammation will have resolved, and the neonate presents with an impalpable testis. Surgical findings include blood vessels and a vas deferens that end blindly at a common point where a small nubbin is occasionally located. Little controversy exists over management of the neonate who was born with a normal scrotal exam but subsequently develops scrotal swelling and erythema; these patients should undergo a Doppler ultrasound and surgical exploration if blood flow cannot be confirmed (Guerra et al, 2008). The persisting controversy surrounds the management of the infant who is born with a clinically apparent torsion. In this setting, immediate exploration in the hours after birth rarely results in testicular salvage, and the risks of anesthesia are elevated. However, multiple case reports have documented bilateral metachronous torsions (Baglaj and Carachi, 2007). Although rare, the potential for bilateral anorchia is devastating given the long-term sequelae of infertility and need for testosterone replacement therapy in puberty and beyond. As the risks of neonatal anesthesia have declined over the years, a growing number of urologists are advocating early exploration with removal of the torsed testes that cannot be salvaged and contralateral orchiopexy to protect the remaining gonad (Guerra et al, 2008). Testes with antenatal torsion may also present as a firm mass in the neonatal period. The use of Doppler ultrasonography will help differentiate testicular torsion from the more rare neonatal yolk sac tumor where testicular blood flow will be demonstrated. In the neonate, this fluid originates in the peritoneal cavity that communicates with the scrotum via an evagination or extension of the peritoneum termed a patent processus vaginalis. This unidirectional fluid flow explains the slow and progressive increase in scrotal size that may be observed in some cases. In other cases with a large patency, the fluid may move in and out of the scrotum with ease such that when the urologist arrives to examine the child, the swelling is absent. It is important to reassure families that communicating hydroceles often improve over time. Indeed, the frequency of a patent processus vaginalis in autopsy series of men with no history of hernias or hydroceles was 20%. In addition, hydroceles may present acutely following the placement of ventriculoperitoneal shunts or peritoneal dialysis catheters. In these instances, both sides should be repaired because there is a 30% chance that the contralateral side will also become symptomatic. However, overly aggressive therapy of the neonatal hydrocele should be avoided, because many of these hydroceles will resolve spontaneously, and surgical intervention may damage the vas deferens or testicular vessels, resulting in testicular atrophy. In determining the proper intervention, it is essential to document the appearance of the scrotum and testes at birth. The newborn who presents in the delivery room with a painless, blue, and edematous hemiscrotum will likely have had an antenatal torsion. This, by definition, is a hernia, which is often easily reducible and not a surgical emergency. In contrast, a difficultto-reduce or incarcerated hernia requires urgent repair to preserve the herniated structure. If the hernia becomes strangulated, blood supply to the intestine will be compromised. In addition, there is an asymmetrical or dorsally hooded foreskin, and the penis may be tethered, creating a significant bend (chordee). The etiology of hypospadias remains uncertain, although a genetic contribution is postulated, because a family history is reported in 5% to 10% of cases. More recently, investigators have postulated that environmental exposures to endocrine disruptors provide a significant contribution (Wang and Baskin, 2008). Experimental studies in mice suggest that exposures to high doses of estrogens lead to malformation of the urethral seam in the developing genital tubercle (Yucel et al, 2003), which would provide a mechanism by which endocrine disruptors could lead to hypospadias. Proponents of the endocrine disruptor theory note that the incidence of hypospadias has increased over the past 30 years (Paulozzi et al, 1997); however, other studies have failed to demonstrate such an increase (Fisch et al, 2009). These infants must not undergo neonatal circumcision; the foreskin is crucial for use in the urethral reconstruction and correction of the chordee. However, up to 10% of patients with a hypospadias may have an intact foreskin, and the diagnosis is determined only after circumcision. In such cases, the loss of foreskin does not compromise the hypospadias repair, which is completed at approximately 6 months of age (Snodgrass and Khavari, 2006). It is important to confirm that both testes are properly descended; hypospadias in conjunction with an undescended testis should raise the possibility of disorder of sexual differentiation (Cox et al, 2008; Kaefer et al, 1999a). This evaluation is critical if both testes are undescended and impalpable, because this phenotype may represent a diagnosis of congenital adrenal hyperplasia in a female neonate (see later section on ambiguous genitalia). For an infant with hypospadias and bilaterally descended testes, an outpatient urological evaluation should be arranged within 2 to 6 months of age. The greatest chance for an underlying disorder of sexual differentiation being present is seen in proximal hypospadias associated with one or two impalpable testes (Kaefer et al, 1999a). The best functional and cosmetic results are obtained in 90% of cases in which the meatus is present anywhere along the penile shaft. Snodgrass and Yucel (2007) have developed a versatile repair that offers high success rates for hypospadias when the meatus is present along the penile shaft. Current hypospadias repairs offer functional improvement in terms of the quality of stream and cosmetic results that are increasingly approaching those seen in normal males. Even the 10% of cases that constitute the most severe penoscrotal hypospadias may be reconstructed with good cosmetic and functional results. The key in all of these cases is the preservation of all genital skin that is to be used in reconstruction-hence the need to avoid circumcision. Circumcision has been practiced for centuries, and despite efforts to discourage the practice, families have continued to request this surgical procedure. In many parts of the United States, nearly 80% of the male population is circumcised. In fact, the incidence of circumcision appears to be rising in the United States (Nelson et al, 2005). In patients with poor hygiene, a nonretractile foreskin may increase the risk of penile cancer and mask the onset of carcinoma of the penis. Yet, penile carcinoma is a rare malignancy in the United States and a common malignancy in underdeveloped nations where circumcision is not practiced. The risk for penile cancer is associated with limited access to regular bathing, basic hygiene, and the ability to retract the foreskin when sexual activity begins. The etiology of carcinoma of the penis is polyfactorial, and the threat of malignancy is not a mandate for circumcision. Although the role of circumcision in Africa is expected to increase given these data, educational efforts on behalf of diminished promiscuity, condom use, and basic hygiene are essential to public health. Parents may cite a significant study reported by Wiswell and Geschke (Wiswell, 1997; Wiswell and Geschke, 1989), that identified a small but very real medical benefit to circumcision. Army hospitals worldwide, these investigators correlated the status of the foreskin with subsequent urinary tract infections. Their findings reveal that, within the 1st year of life, although only 20% of males were not circumcised, this 20% accounted for 80% of all urinary tract infections. In other words, these investigators demonstrated that the presence of foreskin increased the risk of a urinary tract infection by approximately fourfold during the 1st year of life. Therefore, circumcision is to be encouraged for any neonate with an underlying congenital anomaly of the urinary tract such as reflux, posterior urethral valves, or megaureters, where the added burden of a urinary tract infection has serious consequences. The three main techniques for circumcision are those using the Plastibell, the Gomco clamp, and the Mogen clamp. These cases represent the megameatusintact prepuce variant, in which circumcision is not at all detrimental to future reconstruction. C, this photograph shows a more typical hypospadias with an asymmetric foreskin and a meatus that is present at the glanular margin. In such cases, circumcision should be deferred because the hooded preputial skin may be required for subsequent reconstruction. This webbing or tethering can only be repaired by a surgical approach (B); such a patient is not a candidate for circumcision done in the neonatal unit. If there is doubt about the penile articular anatomy, the procedure should be terminated and urologic consultation obtained. Every effort should be made to perform circumcision in the newborn period and thus avoid the use of a general anesthetic in the older child. A study within the Kaiser health plan considered the issue of circumcision from a cost-effectiveness standpoint and came to the conclusion that the system could demonstrate a cost savings of $183 per patient even if routine neonatal circumcision was performed at an average cost of $200 (Schoen et al, 2006). In the child with a complex medical course, this timing also offers the physician a chance to perform a circumcision with full cardiovascular and respiratory monitoring that is not available in an office-based setting. In recent years there has been an increase in the diagnosis of penile webbing, which often results in circumcision being deferred to specialty care and performed under a general anesthetic. Bleeding after a routine circumcision may be observed in 10% to 20% of cases, and minor bleeding is not cause for alarm. If the bleeding has saturated the dressing and repositioning of the pressure dressing fails to stop the bleeding, the frenulum should be inspected because most bleeding vessels are found in this area. Rarely, there is injury to the glans and urethra that requires urological intervention. Typically, the most common problem arising after circumcision is the formation of penile adhesions. This complication may be prevented by teaching parents to retract the penile shaft skin and apply petrolatum over the glans. Parents also should be warned that despite a nice skin fit in the neonatal period, the cosmetic appearance of the penis may change as the prepubic fat pad grows. The result may be that a perfect circumcision is obscured as the penis is pulled back into the fat pad; in such cases, reassurance, and not surgical revision, is indicated. Currently, prenatal diagnosis has allowed discussions about genital ambiguity to begin before delivery (Rintoul and Crombleholme, 2002). The widespread use of ultrasonography and increasing use of amniocentesis have allowed the in utero diagnosis of virilizing congenital adrenal hyperplasia and androgen resistance syndromes. Such diagnosis is possible because the sonographic appearance of genitalia at birth does not correspond to the karyotype obtained at amniocentesis. The most common presentation is the female neonate with congenital adrenal hyperplasia. This virilization is caused by enzymatic deficiencies in the pathways of cortisol synthesis, leading to shunting into the androgen biosynthetic pathways (Speiser and White, 2003). Family history is useful, because an older sibling or relative may have been diagnosed with this condition. Virilizing congenital adrenal hyperplasia is a rare diagnosis, and most neonates with bilateral impalpable testes will prove to be normal males. However, a delay in establishing this diagnosis is dangerous because replacement therapy with cortisol must be initiated. Whereas 20 years ago, surgery was encouraged in the neonatal period with the idea that it would enable better acceptance of a female gender role by the parents and child, today the timing of surgery is controversial; these issues are addressed in the final paragraph. Mutations in the androgen receptor or in the downstream pathway result in a phenotypic female with bilateral testes.
Longterm problems may include spastic diplegia and visual and auditory impairment symptoms 5dpo order on line lopinavir. It appears that the late oligodendrocyte progenitors that populate human cerebral white matter during the high-risk period for white matter injury are the major target of ischemic treatment 3 cm ovarian cyst order lopinavir 250 mg with amex, free radical adhd medications 6 year old proven lopinavir 250 mg, or cytokine injury (Back et al symptoms 8 days before period buy lopinavir master card, 2001 medicine z pack lopinavir 250 mg online, 2002, 2007). However, recent studies show that injury may be due to lack of oligodendrocyte maturation, resulting in decreased myelination and subsequent axonal injury (Segovia et al, 2008). Some data suggest vulnerability of the subplate neurons as well (McQuillen et al, 2003). Tissue damage is associated with proliferation of astrocytes and microglial cells in areas of subcortical degeneration. Increasingly, injury to the developing gray matter is also being identified in the setting of white matter injury and is recognized both in neuropathologic studies and with advanced imaging techniques (Inder et al, 2005; Pierson et al, 2007). The mechanisms of neuronal injury are likely to be similar to those causing injury of the white matter and the preoligodendrocytes. Etiology Hypoxic-ischemic brain injury results when the decrease in cerebral perfusion is severe enough to overwhelm the ability of tissue to extract oxygen from the blood, thereby leading to a mismatch between cerebral blood flow and oxidative metabolism. Although the final pathways leading to cerebral brain death are remarkably similar regardless of the instigating event, the extent, location, and evolution of cell death probably are determined by the nature of the insult. In a study of risk factors for neonatal encephalopathy, antepartum risk factors such as maternal hypotension, infertility treatment, and thyroid disease were present in 69% of cases; both antepartum and intrapartum risk factors were present in 24%; and a history of an intrapartum event such as maternal fever, difficult forceps delivery, breech extraction, cord prolapse, or abruptio placentae was present in 5% of cases (Badawi et al, 1998). A repeat ultrasound examination is recommended between 36 and 40 weeks of postmenstrual age (Ment et al, 2002c). Symptoms usually evolve over a period of 72 hours (Sarnat and Sarnat, 1976) (Table 61-2). During the first 12 hours after birth, the signs and symptoms are secondary to cerebral hemisphere depression, although signs of brainstem involvement may be present. This alteration in consciousness is attributed to the involvement of the cerebral hemispheres, the reticular activating system, or the thalamus. The infant has intact pupillary responses and may have spontaneous eye movements, depending on involvement of cranial nuclei 3, 4, and 6. Cerebral cortical or cerebellar cortical involvement may manifest as hypotonia with decreased movement or as jitteriness or seizure activity, which is seen in 50% of severely affected infants by 6 to 12 hours after birth. In cases of severe asphyxia, seizures may be seen within 2 to 3 hours following the insult. Subtle seizures may also manifest as ocular movements such as tonic horizontal deviation of the eyes or sustained eye opening or blinking; orolingual movements such as tongue or lip smacking or sucking, or rowing or bicycling movements of the extremities, or as recurrent apnea (Mizrahi and Kellaway, 1987). Brainstem release phenomena are common in severely asphyxiated newborns and may be misinterpreted as seizures. During the 12- to 24-hour period after the injury, there is an apparent increase in the level of alertness, but this is not associated with other signs of improvement in neurologic function. This period is accompanied by seizures in 15% to 20% of infants, apneic episodes in 50%, and jitteriness as well as weakness in the proximal limbs in 35% to 50%. Infants who survive until 72 hours continue in stupor with disturbed suck, swallow, and gag reflexes; hypotonia; and weakness of the proximal limbs and especially facial and bulbar musculature. There is sometimes a history of intrauterine distress, as evidenced by abnormalities on the fetal heart tracing, passage of meconium, or a history of difficult labor or delivery with a decrease in placental or fetal blood flow. Excessive jitteriness, seizures, apneic episodes, or abnormal cry are signs of injury. Metabolic complications such as hypoglycemia, hypocalcemia, hyponatremia, hypoxemia, and acidosis are frequently seen. Lumbar puncture should be used to measure cell count, protein and glucose levels, and ratio of lactate to pyruvate, as well as brain-specific creatine kinase. Urinary lactate-to-creatine ratios have been shown to be elevated in asphyxiated infants (Huang et al, 1999). Elevated urinary S100B protein may also serve as a marker of severe perinatal asphyxia (Gazzolo et al, 2004). This is followed after 24 hours by a periodic pattern that consists of periods of greater voltage suppression interspersed with bursts of sharp and slow waves. Subsequently, a "burst suppression" pattern with fewer bursts and more severe voltage depression is seen, followed by an isoelectric tracing. Absence of signal in the posterior limb of the internal capsule is strongly associated with poor neurologic outcome (Rutherford et al, 1995). Ultrasonography is extremely useful for imaging the unstable patient if performed with high-resolution transducers (de Vries et al, 1997). Injury to the basal ganglia, periventricular echodensities, and presence of focal or multifocal ischemic parenchymal lesions suggest neurologic deficits. Ultrasound is insensitive to cortical damage; therefore, in term infants, ultrasound findings may underrepresent the extent of the lesion. It can be used at the bedside to provide information regarding cerebral oxygen delivery. Patterns of Injury and Pathology the mechanism of injury can be one of several types. Different patterns of injury may result, depending on the duration and severity of the insult. Global hypoxia-ischemia develops when the oxygen requirements for cerebral metabolism are unable to be met by cerebral perfusion pressure, as is seen with a decrease in cerebral arterial pressure or an increase in cerebral venous pressure. Although the injury may be transient, it sets into motion a cascade of events that ultimately lead to neuronal death. The duration of the insult necessary to produce brain injury is inversely proportional to the gestational age of the fetus. The extent and location of the injury can be patchy bilaterally, or diffuse and involve the entire cortex. The latent period to neuronal cell death produced by a global insult can range from hours to days and is determined by a complex interaction among various vascular, cellular, and metabolic factors (see later). In some circumstances, lesions may involve the brainstem (pontosubicular necrosis) or the cerebellum. Infants with pontosubicular necrosis have impairment of the cranial nerve nuclei and will exhibit ptosis and facial diparesis. Focal ischemia occurs when the arterial or venous blood supply to a region is compromised. The injured region typically consists of a central dense region of ischemia that undergoes rapid cell death-the core-surrounded by a region of evolving cell injury- the penumbra. The cells in the penumbral region initially are sustained by anaerobic glycolysis. However, within hours the injury becomes irreversible, thus bringing the penumbra into the gradually enlarging area of infarction. This period of delay in cell death provides an opportunity for therapeutic intervention and prevention of further cell damage. Partial asphyxia without acidosis, as occurs in response to impairment of placental gas exchange by uterine contractions, maternal hypotension, or impairment in maternal placental circulation, can produce either widespread cerebral cortical necrosis or a more focal injury to the posterior parietal parasagittal regions. Focal and multifocal cerebral necroses occur secondary to an embolus or thrombus (see Perinatal Stroke, later). There are often underlying problems such as thrombophilias, maternal idiopathic thrombocytopenic purpura, vascular maldevelopment, or history of maternal cocaine use. Infants may present with unilateral, focal seizures or display asymmetric motor function, or they may be asymptomatic. Selective neuronal necrosis can involve specific regions of the cortex, thalamus, brainstem, cerebellum, or hippocampus. An acute global injury such as is seen with uterine rupture or cord prolapse causes injury to the basal ganglia, thalamus, and brainstem, whereas a prolonged partial injury affects mainly the cortex and subcortical white matter. Damage is more extensive in the posterior parietal-occipital region than in the anterior cortical areas. Seizures are a feature of cortical injury, whereas irritability, posturing, and brainstem dysfunction are seen with infarcts affecting the basal ganglia and thalamus. Long-term sequelae of such an injury include cerebral atrophy and multicystic encephalomalacia. Status marmoratus refers to the marbled appearance of the basal ganglia, thalamus, and cerebral cortex in response to the injury. This appearance results from gross shrinkage of the striatum and defects in myelination. It may relate to the type of insult or the density of glutaminergic receptors in the basal ganglia (Johnston, 1995). Although the clinical correlate of this condition in the newborn is not well defined, it is seen in children with choreoathetoid cerebral palsy. Cerebral blood flow is closely autoregulated over a wide range of systemic blood pressure by either vasoconstriction or dilatation of cerebral arterioles. Therefore, rapid changes in cerebral perfusion pressure can occur in response to changes in systemic blood pressure, as well as changes in cerebral Pao2 and Paco2. Injury typically tends to occur in the watershed regions, which in the term newborn are located in the parasagittal regions of the cerebral cortex. The periventricular white matter is the most vulnerable region in preterm newborns. Clinical features of parasagittal injury include hypotonia and weakness, especially in the upper trunk. Cortical infarcts usually occur in the watershed region supplied by the most peripheral branches of the anterior, middle, and posterior cerebral arteries. In patients with hyporeflexia or areflexia, spinal cord pathology should be considered. Although overall cerebral O2 demands are lower in the infant than in the adult, cerebral oxidative metabolism is considerably increased in areas of active neural development that are associated with either synapse formation or activation of enzymes required for ion homeostasis. Glucose is the primary source of energy in cerebral metabolism, and although the newborn brain is capable of utilizing alternative energy substrates such as ketones, lactate, and free fatty acids, glucose uptake mechanisms are relatively underdeveloped (Cremer et al, 1979; Gregoire et al, 1978). Animal studies have shown that this impaired uptake of glucose can impair cerebral metabolism even before oxygen depletion (Yager et al, 1996). Vasoautoregulation in response to increased cerebral blood pressure or flow is relatively underdeveloped in the newborn, thus rendering the infant more vulnerable to ischemic events. The gradual increase in vascularity of the developing brain leads to the creation of watershed areas. These global events lead to patterns of focal injury that are dependent on maturational processes. The similarities between processes essential for brain development and those mediating cellular injury make the immature brain particularly vulnerable to ischemic insult. These similarities include an increased density of glutamate receptors, an increase in glutaminergic synapses in particular regions of the immature brain, and enhanced accumulation of cytosolic calcium after activation of the glutamate receptor. It has been shown that there are proportionately more glutamate receptors in the immature rat brain than in the mature rat brain and that the developing rat brain is much more sensitive to injury than the newborn or adult brain (Yager et al, 1996). This is followed by neuronal membrane depolarization and release of neurotransmitters such as glutamate, which increase cytosolic calcium and induce destructive enzymes and free radicals. Reoxygenation after the ischemic episode plays a significant role in cellular injury. In the normal state, glucose and oxygen are the main requirements for brain energy production, which occurs by oxidative phosphorylation. Glucose is taken up by a carrier-mediated diffusion process and phosphorylated to glucose 6-phosphate, the major portion of which enters the glycolytic pathway to form pyruvate. A failure of the pump leads to an influx of sodium into the cell and potassium outside the cell. Associated glial uptake of sodium and water leads to astrocytic swelling that in turn decreases diffusion of oxygen and glucose to the neurons. Accumulation of lactate secondary to anaerobic glycolysis leads to tissue acidosis, which inhibits both vascular autoregulation and phosphofructokinase, the rate-limiting enzyme in glycolysis. In immature animals, hypoglycemia has been shown to be damaging; pretreating animals with glucose decreases the impact of the injury when given before, but not during, the injury (Sheldon et al, 1992; Vannucci and Vannucci, 2000). Excitotoxicity refers to excessive glutamatergic activation that leads to cell injury and death (Olney, 2003). Neuronal injury is initiated by release of glutamate and other excitatory neurotransmitters from the presynaptic neurons. Subsequently, glutamate release occurs by both a reversal of normal glutamate uptake mechanisms by the nerve terminals and glia, and membrane leakage. The density of receptors is higher in regions of active development, and the different subtypes vary in different regions of the brain at different gestational ages. Activation of any of the three subtypes of glutamate-activated postsynaptic neuron receptors leads to an influx of calcium into the postsynaptic neurons. Activation of the metabotropic receptor results in the generation of inositol triphosphate, which triggers release of sequestered calcium. The increase in intracellular calcium sets into motion an irreversible cascade of events that leads to cell injury. Calcium activates several degradative enzymes such as phospholipases, proteases, and endonucleases. Activated phospholipases such as phospholipase A2 hydrolyze membrane phospholipid, thereby releasing free fatty acids such as arachidonic acid. Enzyme activation as well as activation of xanthine and prostaglandins generates free radicals that perpetuate the injury by lipid membrane peroxidation. Iron is usually maintained in a nontoxic "ferric" state but is reduced into the injurious "ferrous" form, which can react with oxygen-reactive species to propagate further injury. During the initial period of reperfusion there is a clearance of glutamate (Takahashi et al, 1997). Generation of free radicals during this phase occurs by two methods: free fatty acids enter the cyclooxygenase pathway and generate arachidonic acid and prostaglandins, and xanthine oxidase converts hypoxanthine to uric acid. Free radicals activate adhesion molecules in platelets and leukocytes, which increases occlusion of the microvasculature, thereby perpetuating injury. Apoptosis refers to programmed cell death, a mechanism that is ongoing during the process of brain maturation. Necrotic cells are characterized by cellular swelling, fracture of cell membranes, and an inflammatory cellular reaction.
Lopinavir 250 mg with amex. Treatment For Pneumonia - Treatment And Prevention Of Pneumonia.
Unless no other choice is available treatment kidney failure cheap 250 mg lopinavir with visa, use of the subclavian artery should be avoided in infants who are likely to require longterm renal replacement therapy fungal nail treatment purchase lopinavir 250 mg on line, because in the future the forearm fistula of the ipsilateral arm can fail with mild stenosis of the subclavian vein medicine ball workouts buy generic lopinavir 250 mg on-line. Peritoneal Dialysis Once a peritoneal dialysis access catheter is placed and the decision to start dialysis has occurred treatment authorization request purchase genuine lopinavir on line, small-volume continuous cycles (10 mL/kg) are performed medications contraindicated in pregnancy lopinavir 250 mg purchase with mastercard. Fluid is filled with dialysate solution, left in the peritoneal cavity to dwell, and drained. The dextrose concentration in the fluid will determine the amount of net water losses (ultrafiltration). Complications associated with peritoneal dialysis include peritonitis, leakage around the catheter exit site rendering the dialysis virtually impossible, tunnel infection, catheter malfunction, and obstruction by omentum (Coulthard and Vernon, 1995). Fluid leakage into other compartments (including the chest in patients without an intact diaphragm) can occur and if suspected, the fluid composition will reveal high glucose levels if a leak is present. Absolute or relative contraindications to peritoneal dialysis include necrotizing enterocolitis, abdominal wall defects, and the presence of an intraabdominal foreign body, such as a ventriculoperitoneal shunt or diaphragmatic patch. Hemodialysis Once reliable access to the vascular space is achieved, the hemodialysis procedure can be performed in neonates. Because much larger volumes of dialysis are used, the blood flow becomes the limiting factor in the amount of clearance that can be achieved. Even with the smallest dialyzers and neonatal tubing, most infants need blood priming of the extracorporeal circuit for therapy. Skilled pediatric hemodialysis nurses are required at the bedside during the entire procedure, which typically lasts 3 to 4 hours. Achieving adequate fluid removal is sometimes difficult, especially in hemodynamically unstable infants. This approach usually requires systemic heparinization, with activated clotting time usually maintained at 180 to 200 seconds, rendering this approach risky in preterm newborns and others at high risk for intracranial bleeding. The advent of newer roller pump technology improved the accuracy of small flows as low as 10 mL/min and provided the option of using the machine to pump blood via a double lumen catheter placed in a major vein. The main advantage of a continuous modality is that lower blood flow and fluid removal rates can be used to accomplish the desired ultrafiltration and clearance goals. The advantage of regional citrate anticoagulation is that the patient is not anticoagulated; however, this method of anticoagulation has the added risk of hypocalcemia caused from citrate excess (especially in those infants with impaired liver metabolism) and metabolic alkalosis (Tolwani and Willie, 2009). Approximately 8% (35 neonates) in the registry were dialyzed in the 1st month of life. Of the 35 infants in the registry, 24 received dialysis for fluid overload, electrolyte imbalance, or both, and 11 of 35 received dialysis for inborn errors of metabolism. Infants receiving dialysis for inborn errors had a better survival rate (73%) compared with others (30%). In addition, because standard protocols on citrate regional anticoagulation anticipate normal liver metabolism of citrate, caution must be exercised when performing dialysis in premature infants or newborns with multiple organ failure who may have impaired liver function. This compensatory hypertrophy causes glomeruli to function under increased intracapillary hydraulic pressure, which over time causes damage to capillary walls. The hyperfiltration hypothesis has been applied and confirmed in autopsy data of patients with hypertension (Keller et al, 2003; Ohishi et al, 1995) and has been reported at length in infants with intrauterine growth retardation (Barker and Osmond, 1988; Barker et al, 1989; Manalich et al, 2000; Wadsworth et al, 1985; White et al, 2009). A systematic review and metaanalysis in 2009 concluded that low-birthweight infants (5. Premature infants (even those born appropriate for gestational age) are born with low nephron numbers. Using computer-assisted morphometry, Rodriguez et al (2004) showed that premature infants have lower numbers of nephrons compared with term infants. Premature infants who had long survival (to at least 36 weeks after conception) had nephron numbers similar to those in premature infants with short survival, suggesting that the extrauterine environment does not allow for proper neoglomerulogenesis. Animal models suggest that although tubular recovery occurs, damage to vascular endothelial cells remains and leads to interstitial fibrosis and progressive kidney dysfunction (Basile et al, 2001). Rees (2008) reported that six new infants per 1 million population initiate dialysis per year compared to three per 1 million in the United Kingdom. They also found that 53% of these infants were premature, a figure significantly higher than in the total infant population of Germany. The most common causes of renal failure in most studies of neonates are renal dysplasia or obstructive uropathy (Carey et al, 2007; Ledermann et al, 1999; Rees, 2008; Shroff et al, 2003; Wedekin et al, 2008)-specifically, posterior urethral valves (Ledermann et al, 1999). According to the guidelines, anemia is defined as a hemoglobin (Hgb) concentration less than the 5th percentile of normal for age and sex (Table 85-4). The normative values used to define anemia in children older than 1 year are taken from the third National Health and Nutrition Examination Survey (Astor et al, 2002) database, whereas the norms for infants younger than 1 year are derived from other reference sources (Nathan and Orkin, 2003). Other potential contributing factors include a shortened red blood cell life span, secondary hyperparathyroidism, hypothyroidism, folate and vitamin B12 deficiency, chronic inflammation and hemoglobinopathies. The etiology of absolute iron deficiency is multifactorial and can be related to poor intake, gastrointestinal blood loss, and repeated phlebotomies for laboratory tests. Iron therapy typically consists of the provision of oral elemental iron in doses ranging from 2 to 3 mg/kg/day up to 6 mg/kg/day in two to three divided doses (National Kidney Foundation, 2006). Iron should be taken 2 hours before or 1 hour after all calcium containing phosphate binders to maximize gastrointestinal absorption. In turn, the rate of rise of the Hgb level should be no more than 1 to 2 g/dL per month. Metabolic acidosis leads to both endogenous growth hormone and recombinant growth hormone resistance. The World Health Organization Growth Standards of length-for-age, weight-forage, weight-for-length, body mass indexfor-age and head circumferencefor-age should be used as the reference for children from birth to 2 years (World Health Organization, 2006). Energy requirements should, however, be considered to be 100% of the estimated energy requirement for chronologic age (Table 85-6) (Food and Nutrition Board, 2002; Ruely et al, 1989). Supplemental nutritional support is indicated when the voluntary intake by the child fails to meet energy requirements and the child is not achieving expected rates of weight gain or growth for age. The development of repeated emesis in children fed per nasogastric tube has prompted the use of gastrostomy as the preferred route of therapy (Warady et al, 1996). In infants younger than 1 year, an initial infusion rate of 10 to 20 mL/h or 1 to 2 mL/ kg/h is generally well tolerated, to be followed by a daily increase of 5 to 10 mL per 8 hours or 1 mL/kg/h toward achieving the treatment goal. At the same time, there is no evidence that strict dietary protein restriction (120% recommended daily allowance) has any nephroprotective effect, nor does this level of intake compromise growth. In patients receiving dialysis, the protein requirements are increased to account for dialysis-related protein losses. This scenario can result in contraction of the extracellular volume, in addition to having an adverse effect on growth and nitrogen retention (Wassner and Kulin, 1990). The same holds true for infants receiving peritoneal dialysis even if they are anuric, because most patients lose significant quantities of sodium in the dialysate. In patients who remain hyperkalemic despite repletion of salt and water, restriction of dietary potassium intake is critical. For infants and young children, 40 to 120 mg (1 to 3 mmol/kg/d) of potassium may be a reasonable start. Breast milk has a lower potassium content (546 mg/L; 14 mmol/L) than commercial milkbased infant formula (700 to 740 mg/L; 18 to 19 mmol/L) (National Kidney Foundation, 2009). Pretreatment of infant formula with a potassium binder, treatment of constipation, and attention to medications that can exacerbate hyperkalemia. Ethics of Initiating or Withdrawing Renal Replacement Therapy Decisions to withdraw or withhold treatment have to be made for many patients in neonatology units, and for as many as 30% to 58% of patients in pediatric intensive care units. With the advent of advanced technology in the 1980s that made possible the provision of safe and effective peritoneal dialysis to even the smallest infant came increasing ethical dilemmas and significant variations in practice (Fauriel et al, 2004; Shooter and Watson, 2000; Watson and Shooter, 2004). Evidence for this variation in practice has been seen in other surveys as well (Fauriel et al, 2004). Most clinicians agree that there is more to their skill than the indiscriminant application of technology. In the end, clinicians and parents often struggle bravely to reach a compassionate decision with as much agreement as possible. Principles of practice that may provide valuable assistance in this process have been published previously (Watson and Shooter, 2004). Renal Replacement Therapy End-stage renal disease is an uncommon disorder in children, with an incidence in the United States of approximately 14 patients per 1 million children of a similar age (U. The incidence varies within the pediatric population with a rate of 29 per 1 million for children 15 to 19 years old, in contrast to a rate of 9 per 1 million for children 0 to 4 years old. Peritoneal dialysis makes use of the peritoneal membrane as a natural dialyzing membrane. Dialysis solution is instilled and dwells within the peritoneal cavity, during which time bloodstream-derived solutes move down a concentration gradient based on diffusion, and fluid is removed as a result of the osmotic gradient created by the dextrose component of the dialysis fluid. The inflow, dwell, and drainage of dialysate characterize a single dialysis cycle or exchange. The incidence of this infection is greatest during infancy, with a rate of 1 infection every 14. Whereas gram-positive organisms account for the majority of infections, gram-negative episodes of peritonitis are common in infants and young children (Zurowska et al, 2008). In turn, when peritonitis is suggested, empiric antibiotic therapy should provide coverage for gram-positive and gram-negative organisms (Warady et al, 2000). In some cases, infants may experience hypogammaglobulinemia in this situation and may benefit from replacement therapy (Neu et al, 1998). Other treatment-related complications that occur most frequently during infancy include anterior ischemic optic neuropathy and sudden blindness secondary to hypovolemia, excessive loss of protein across the peritoneal membrane and hernia formation (Lapeyraque et al, 2003; Quan and Baum, 1996; Warady et al, 2009). The procedure is complicated, and limited clinical experience has revealed a high incidence of patient morbidity (Al-Hermi et al, 1999; Kovalski et al, 2007; Shroff et al, 2007). Blood access is typically in the form of a 7F or 8F dual-lumen catheter, which allows blood pump flow rates of 30 to 50 mL/min. The complicated nature of the procedure mandates that it be performed only in highly qualified centers. In short, transplantation is a viable alternative for these young patients and is their best hope for long-term survival. Minimizing the use of interventions that greatly increase the risk of central venous thrombosis should also be encouraged. Renal Data System reveals that 51 infants younger than 1 year and 89 children aged 1 to 4 years received a kidney transplant in 2006 (U. The 3-year graft survival for patients who received a living donor transplant in 2003 was 83. In the case of deceased donor transplants, the graft survival for those aged 1 to 4 years was 89. Protein-calorie malnutrition, metabolic acidosis, electrolyte disarray, renal osteodystrophy, and changes in the gonadotropic hormone axis in the face of uremia, corticosteroid treatment, or both are factors that contribute to this challenging problem (Geary, 1998; Haffner, 2008). Several studies have shown that infants have impaired growth at initiation of chronic dialysis. There are conflicting reports regarding improvement in growth with renal replacement therapy. Most studies suggest that young children on dialysis fail to grow well, despite meeting 100% of the recommended daily allowance of caloric intake (Shroff et al, 2003). Recent reports have described improved longitudinal growth and sustained catch-up growth in infants with chronic renal failure in whom growth hormone treatment was initiated in the 1st year of life. Mencarelli et al (2009) reported 12 infants initiated on recombinant human growth hormone at a median age of 0. The brain undergoes rapid growth during infancy, reaching half of its adult weight by 6 months of age (Harris, 2006). Postnatal brain growth includes neuronal differentiation, dendritic branching, and axonal myelination (Gipson, 2008). Renal impairment in infancy, a crucial time of neural development, raises concerns regarding the neurodevelopmental outcomes in these children. In one study, 28 patients initiating chronic peritoneal dialysis by 3 months of age underwent formal neurodevelopmental testing. Fifteen of the 16 school-aged patients were fulltime students in age-appropriate classrooms, and all of the children younger than 5 years were in preschool (Warady, 1999). Ten (67%) children had an intelligence quotient in the normal range, whereas 13 of 15 (87%) were within 2 standard deviations of the mean (Madden et al, 2003). Factors reportedly associated with mortality include African American race, presence of comorbidities such as chronic lung disease, multiorgan dysfunction, diagnosis of a syndrome, and oliguria or anuria (Hijazi et al, 2009). Approximately 25% of neonates died over 18 months of follow-up in one study, which was similar to the mortality rate of older children (Carey et al, 2007). However, if the patients were receiving renal replacement therapy, survival improved to 91%, 83%, and 83% for 1, 2, and 5 years, respectively. Among the patients who received renal transplants, the 1-, 2-, and 5-year survival rates were greater than 95% (Wedekin et al, 2008). It appears that younger children are receiving transplants more readily in recent years (Carey), which is encouraging in the face of more favorable survival. In one study of 18 children requiring chronic hemodialysis by 2 years of age, the median number of hospital admissions while receiving dialysis was 6 (range 3 to 16). Another study divided 698 children requiring chronic dialysis by 2 years old into those initiating dialysis by 1 month of age and those initiating dialysis between 1 month and 24 months of age. Approximately 80% of children in both groups required hospitalization at some point in the 13-year follow-up period. Among children ever hospitalized, those initiating dialysis as neonates were hospitalized more frequently than were children starting dialysis later (mean number of hospitalizations, 54 versus 39; p <0. Hijazi R, Abitbol C, Chandar J, et al: Twenty-five years of infant dialysis: a single center experience, J Pediatr 155:111-117, 2009. Rees L: Management of the neonate with chronic renal failure, Semin Fetal Neonat Med 13:181-188, 2008. Shooter M, Watson A: the ethics of withholding and withdrawing dialysis therapy in infants, Pediatr Nephrol 14:347-351, 2000.
Intramural perfusion medicine 832 order genuine lopinavir on line, however medications without doctors prescription 250 mg lopinavir purchase otc, could be adversely affected by the prominent trabeculations and intratrabecular recesses medications such as seasonale are designed to cheap 250 mg lopinavir free shipping, particularly the subendocardium medications 4h2 250 mg lopinavir order fast delivery. The increased fibrous and elastic tissue on the endocardial surfaces could be due to subendocardial ischemia medicine lodge treaty discount lopinavir 250 mg amex, perhaps in response to isometric contraction among the trabeculae and recesses. The neonates who died all had systemic disease (mitochondrial and other metabolic disorders); they demonstrated a 5-year survival rate of 86%. When patients receiving transplants were added, the 5-year survival free of death or transplantation was 75%. More recent reports, however, have shown a more benign natural history, with lower risk for (malignant) ventricular arrhythmias. Histologic examination shows myocardium around deep intratrabecular recesses that can serve as slow conducting zones with reentry. Clinically, this subgroup appears to be benign during childhood and approximates 25% of all subjects. This subgroup is usually followed up yearly in the outpatient clinic, and patients are not treated with medication and or restricted from activities. These patients appear to have an elevated risk of sudden events and require closer follow-up and therapeutic intervention with either medication or implantable defibrillator, depending on the specific arrhythmia and associated symptoms. Hypercontractile systolic function and diastolic dysfunction and appear to have similar outcomes. These young children, particularly infants, can succumb to heart failure or arrhythmias, especially if they have metabolic derangement. In this case, the surgeon, cardiac anesthesiologist, and cardiac intensivist must play close attention to the myocardial function and treat it expectantly. Using the ratio of noncompaction to compaction, the authors analyzed the 16 segments to determine whether severity was correlated with poor outcomes in these affected children. Twenty-eight patients (64%) remained alive (group 1), and 16 patients (36%) either died or were received a transplant (group 2). Similar regions of involvement in the 16-segment model were notable, with sparing of basal segments and involvement of the midpapillary and apical regions (P <. This gene encodes a novel protein family (tafazzins) that participate in the metabolism of cardiolipin, the signature phospholipid of mitochondria, and is also responsible for Barth syndrome and other forms of infantile cardiomyopathies. Congenital heart defects have not been associated with Barth syndrome or other G4. Consistent with X-linked inheritance, no male-to-male transmission of the disease occurs. Therapy and Outcome the specific therapy depends on the clinical and echocardiographic findings. In patients with systolic dysfunction and heart failure, anticongestive therapy identical to those used in patients with dilated cardiomyopathy is appropriate. In particular, angiotensin converting enzyme inhibitors and -adrenergic blocking agents are useful. Diuretics may also be needed; however, in patients exhibiting findings more consistent with a hypertrophic cardiomyopathy or diastolic dysfunction physiologic phenotype, -blocker therapy alone may be more appropriate in children. In adults and a small cadre of at-risk children, primary prevention is commonly considered. Intimate understanding of the cardiac function abnormalities, evidence of thrombi (which should be treated with anticoagulation), and the metabolic status of the patient must be addressed by the interventional cardiologist, cardiac anesthesiologist, and surgeon in approaching these patients invasively. Finsterer J, Stöllberger C, Fazio G: Neuromuscular disorders in left ventricular hypertrabeculation/ noncompaction. Oechslin E, Jenni R: Left ventricular noncompaction revisited: a distinct phenotype with genetic heterogeneity Ichida F, Hamamichi Y, Miyawaki T, et al: Clinical features of isolated noncompaction of the ventricular myocardium: Long-term clinical course, hemodynamic properties and genetic background. Stöllberger C, Blazek G, Dobias C, et al: Frequency of stroke and embolism in left ventricular hypertrabeculation/noncompaction. Steffel J, Kobza R, Oechslin E, et al: Electrocardiographic characteristics at initial diagnosis in patients with isolated left ventricular noncompaction. Caliskan K, Ujvari B, Bauernfeind T, et al: the prevalence of early repolarization in patients with 16. Celiker A, Ozkutlu S, Dilber E, et al: Rhythm abnormalities in children with isolated ventricular noncompaction. Aras D, Tufekcioglu O, Ergun K, et al: Clinical features of isolated ventricular noncompaction in adults long-term clinical course, echocardiographic properties, and predictors of left ventricular failure. Caliskan K, Kardos A, Szili-Torok T: Empty handed: A call for an international registry of risk stratification to reduce the "sudden-ness" of death in patients with non-compaction cardiomyopathy. Stöllberger C, Blazek G, Wegner C, et al: Heart failure, atrial fibrillation and neuromuscular disorders influence mortality in left ventricular hypertrabeculation/noncompaction. Thuny F, Jacquier A, Jop B, et al: Assessment of left ventricular non-compaction in adults: 31. Kobza R, Steffel J, Erne P, et al: Implantable cardioverter defibrillator and cardiac resynchronization therapy in patients with left ventricular noncompaction. Autore C, Quarta G, Spirito P: Risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Klaassen S, Probst S, Oechslin E, et al: Mutations in sarcomere protein genes in left ventricular noncompaction. Tang S, Batra A, Zhang Y, et al: Left ventricular noncompaction is associated with mutations in the mitochondrial genome. Ventricular Arrhythmias in Takotsubo Cardiomyopathy Christopher Madias and Richard G. The exact mechanism of the transient cardiomyopathy remains undefined; however, it is believed that ventricular dysfunction is induced by catecholamine-mediated myocardial toxicity. Myocardial dysfunction most commonly results in apical ballooning; however, other distinct patterns of regional myocardial involvement have been described in a minority of patients, including midventricular and basal distributions. Supraventricular arrhythmias, including atrial fibrillation and supraventricular tachycardia, have also been described in this disorder and are likely attributable in part to high levels of circulating catecholamines. Evidence suggests that a genetic predisposition might increase the risk of arrhythmic death in acute coronary events. An understanding of the clinical circumstances associated with ventricular arrhythmias is also critical. Among other therapies, potassium supplementation to maintain serum concentrations at high-normal levels is suggested for management of drug-induced TdP. In light of evidence indicating catecholamine excess in the genesis of this syndrome, temporary pacing should likely be favored over intravenous isoproterenol. Abe Y, Kondo M, Matsuoka R, et al: Assessment of clinical features in transient left ventricular apical ballooning. Kurisu S, Inoue I, Kawagoe T, et al: Time course of electrocardiographic changes in patients with tako-tsubo syndrome: Comparison with acute myocardial infarction with minimal enzymatic release. Mitsuma W, Kodama M, Ito M, et al: Serial electrocardiographic findings in women with Takotsubo cardiomyopathy. Dote K, Sato H, Tateishi H, et al: [Myocardial stunning due to simultaneous multivessel coronary spasms: A review of 5 cases]. Tsuchihashi K, Ueshima K, Uchida T, et al: Transient left ventricular apical ballooning without coronary artery stenosis: A novel heart syndrome mimicking acute myocardial infarction. Eitel I, von Knobelsdorff-Brenkenhoff F, Bernhardt P, et al: Clinical characteristics and cardiovascular magnetic resonance findings in stress (takotsubo) cardiomyopathy. Kurowski V, Kaiser A, von Hof K, et al: Apical and midventricular transient left ventricular dysfunction syndrome (tako-tsubo cardiomyopathy): Frequency, mechanisms, and prognosis. Yamada Y, Tani T, Homma M, et al: Two cases of torsades de pointes associated with Takotsubo cardiomyopathy as the second insult. Bonnemeier H, Demming T, Weidtmann B, et al: Differential heart rate dynamics in transient left ventricular apical and midventricular ballooning. Iga K, Gen H, Tomonaga G, et al: Reversible left ventricular wall motion impairment caused by pheochromocytoma-a case report. Matsuoka K, Okubo S, Fujii E, et al: Evaluation of the arrhythmogenicity of stress-induced "Takotsubo cardiomyopathy" from the time course of the 12-lead surface electrocardiogram. Thakar S, Chandra P, Hollander G, et al: Electrocardiographic changes in Takotsubo cardiomyopathy. Kosuge M, Ebina T, Hibi K, et al: Simple and accurate electrocardiographic criteria to differentiate takotsubo cardiomyopathy from anterior acute myocardial infarction. Kosuge M, Ebina T, Hibi K, et al: Differences in negative T waves between takotsubo cardiomyopathy and reperfused anterior acute myocardial infarction. Nakajima T, Kagoshima T, Fujimoto S, et al: the deeper the negativity of the T waves recorded, the greater is the effectiveness of reperfusion of the myocardium. Kurisu S, Kato Y, Mitsuba N, et al: Comparison of electrocardiographic findings between the midventricular ballooning form and the apical ballooning form of takotsubo cardiomyopathy. At least 13 different genetic variants of the syndrome are known nowadays, with more than 300 mutations reported. The extremely wide genetic heterogeneity of inherited arrhythmia disorders, along with the sometimes almost impossible genotypeto-phenotype correlations, the overlapping electrophysiological manifestations, and the difficulties associated with diagnosis and assessment of prognosis, makes this new field of genetic rhythmology a fascinating one. In this chapter we review present knowledge, progress that has been made, and future research directions for Brugada syndrome. The second Brugada Syndrome Consensus Report of 2005 (endorsed by the Heart Rhythm Society and the European Heart Rhythm Association)2 stated the current recommendations regarding diagnostic criteria. On the basis of the results of comparative studies5 and clinical experience, ajmaline, at a dose of 1 mg/kg, is the best drug. Some patients present with syncope or (aborted) sudden cardiac death caused by malignant ventricular arrhythmias; however, others are completely asymptomatic, and no apparent structural heart disease can be found. It has been suggested that the embryologic origin of the right ventricle (neural crest cells) differs from that of the left ventricle, and this fact predisposes the right ventricle to arrhythmias in adulthood. The fast transient outward K+ current Ito is most prominent in epicardial cells of the right ventricle. The 2/-subunit of voltage-dependent calcium channels regulates current density and activation/ inactivation kinetics of the calcium channel. It is well known that environment may play a role in the predisposition to arrhythmias in patients with Brugada syndrome. Recent data suggest that the type of genetic mutation can serve as a tool for risk stratification in BrS. In this study, patients and relatives with a truncated protein had a more severe phenotype and more severe conduction disorders. Although this provides proof of the concept that some mutations appear to confer a worse prognosis, use of these data in the clinical setting is not yet sufficient to alter clinical decisions. Administration of antiarrhythmic drugs in these cases can lead to ventricular fibrillation and sudden death. Approximately 15% to 20% of patients with BrS develop supraventricular arrhythmias. These so-called overlapping syndromes represent a tremendous challenge to physicians for diagnosis and risk stratification. To date, some markers of high risk in BrS patients have been clearly identified and accepted by all groups, but the issue of risk stratification of asymptomatic BrS patients remains controversial. The reported annual rate of events has decreased from the time the first patients were reported to the most recent published series; this change probably reflects inherent bias during the first years following the description of a novel disease in which particularly severe forms of the disease are most likely to be diagnosed. Male sex has consistently been associated with a trend toward presentation of more arrhythmic events and even has been defined as an independent predictor of a worse outcome in a meta-analysis. This is the first study that proposed the use of genetics in risk stratification for BrS. The literature includes reports on two types of series: ones with almost no events at all during follow-up, in which obviously the lack of events brings a negative value for any studied factor; and others with a reasonable number of events during follow-up, for which different factors have been studied, some of which have shown value for stratification. The discussion is not about which factor is more efficient for stratifying patients but rather why there is this big and so far unexplained difference among series. Certainly some international consensus will have to be reached on how the diagnosis should be made based on the findings of new and updated studies. Published data suggest that they might play a role in the phenotypic manifestations of BrS. With the hormonal influence hypothesis, the few available data existing thus far of BrS in children have shown no difference in phenotypic presentation between boys and girls. Brugada Syndrome in Children Sudden cardiac death accounts for approximately 20% of sudden deaths in the pediatric age group. In the initial description of the disease, three out of eight patients were children. Moreover, no standardized data are available for optimal positioning of the right precordial leads in children, and the shape of the chest in a growing body can lead to confusion. In contrast to adults, no male predominance is found in symptomatic pediatric patients. This finding could be related to lower levels of testosterone in prepubertal children. Moreover, if it is taken into account that a false-negative result can be seen in up to 30%, depending on which drug is given, the question is whether a second test should be performed some years later. Bradyarrhythmias can be a cause of death in these patients; thus pacemaker implantation is mandatory in certain cases. Genetic testing should be performed in index cases, and, when positive, mutations should be searched in children, whatever age they are, to follow recommendations on fever control and avoidance of listed drugs. Research into stem cells is one of the last fields to be incorporated into the cardiac arrhythmia scenario. Animal models are useful for researchers seeking to understand the role of genetic and environmental modifiers in cardiac electrical activity. A full in silico model of the potassium channel has been developed that shows the available structures of channels, including all transmembrane segments. Future work will be aided by the use of these new tools in the field of biomedicine. Future Research and Direction In recent years, cardiovascular studies have been focused on personalizing risk assessment and determining optimal therapy on an individual basis. However, future research in genetics, epigenetics, transcriptomics, proteomics, metabolomics, and animal model approaches will explore the complexity of BrS-like diseases by establishing and using more reliable models at in silico, in vitro, and in vivo levels. Burashnikov E, Pfeiffer R, Barajas-Martinez H, et al: Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death.
References
- Chapman I: Morphogenesis of occluding coronary artery thrombosis. Arch Pathol 1965;80:256-261.
- Bach T, Geavlete B, Herrmann TW, et al: Retrograde blind endoureterotomy for subtotal ureteral strictures: a new technique, J Endourol 22:2565, 2008.
- Rassweiler, J., Seemann, O., Hatziger, M., Schulze, M., Frede, T. Technical evolution of laparoscopic radical prostatectomy after 450 cases. J Endourol 2003;17:143-154.
- Murphy PG, Grondin J, Altares M, Richardson PM. Induction of interleukin-6 in axotomized sensory neurons. J Neurosci 1995;15:5130-5138.
- Stanson AW, Kazmier FJ, Hollier LH, et al. Penetrating atherosclerotic ulcers of the thoracic aorta: natural history and clinicopathologic correlations. Ann Vasc Surg. 1986;1:15-23.
- Quinones MA, Waggoner AD, Reduto LA, et al. A new, simplified and accurate method for determining ejection fraction with two-dimensional echocardiography. Circulation 1981; 64:744-753.
- Hook GE, Gilmore LB, Talley FA. Multilamelled structures from the lungs of patients with pulmonary alveolar proteinosis. Lab Invest 1984;50:711-25.