

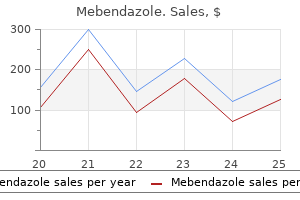
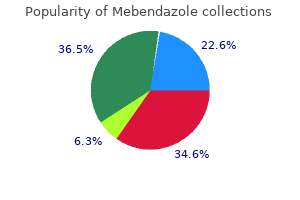
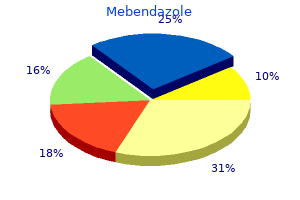
Mebendazole
| Contato
Página Inicial
Edward Christian Healy, M.B.A., M.D.
- Chairman of Cardiology, Suburban Hospital
https://www.hopkinsmedicine.org/profiles/results/directory/profile/2290046/edward-healy
It clearly runs in families anti viral apps mebendazole 100 mg mastercard, but the exact mode of inheritance has not yet been defined antiviral journals mebendazole 100 mg order with amex. A preexisting neurological abnormality and strong family history of epilepsy add complexity to the prognosis antiviral y antibiotico juntos order mebendazole in india. The most common precipitants are viral infections four stages hiv infection order mebendazole discount, and specific association with herpes virus type 6 has been reported hiv infection rate ghana buy mebendazole 100 mg amex. Oral administration of diazepam during febrile diseases to prevent recurrence has been advocated. The subsyndromes of tapping epilepsy, proprioceptive-induced seizures, cortical reflex myoclonus, and startle epilepsy are symptomatic focal epileptic syndromes associated with cerebral static lesions, acute illness, or progressive encephalopathies. Startle epilepsies are more frequent in children, and are especially associated with large cortical lesions in children with congenital hemiplegia. They are characterized by seizures triggered by elaborate stimuli whose specific pattern is the determining factor in seizure evocation. Primary reading epilepsy has been classified as an idiopathic localization-related epilepsy, and thinking epilepsy may be seen as part of an idiopathic generalized epilepsy. All these four complex reflex epilepsies are rare, and prognosis depends on etiology. Early surgery may be attempted in exceptional cases with clear focal cerebral lesions. Clinical manifestations: Onset of seizures takes place during the first 3 months of life, erratic focal myoclonus being the first sign in many instances. Other focal motor seizures are frequent, including brief tonic seizures, but epileptic spasms are rare and may appear later. Marked hypotonia of truncal muscles is present from onset, but hypertonia of limbs may develop. Prognosis: the outcome is consistently unfavorable, and more than half of the patients die during the first 2 years of life. Clinical manifestations: Onset is during the first 6 months of life with focal motor seizures alternating from one hemisphere to the other. Clinical manifestations: Typical infantile spasms in otherwise normal babies usually appear between the 4th and 9th months of life. The most well-known type is flexor, including symmetrical flexor contraction of axial muscle groups plus abduction and semiflection of the upper arms. Most of the spasms occur in clusters, predominantly soon after awakening or upon falling asleep. After only one or several weeks, the baby has less eye contact, and is less responsive in general. Differential diagnosis with nonepileptic conditions appearing in babies is very important, mainly with benign myoclonus of early infancy. Familial cases are rare, and most frequent etiologies are prenatal cerebral lesions or malformations. Clinical manifestations: Onset of seizures is within the first 3 months of life, mainly during the first month. The main seizure type consists of brief tonic spasms, with or without clustering, occurring during awake and sleep states. Focal clonic seizures are observed in some patients, but myoclonic seizures are rare. Multiple types of generalized seizures are characteristic, mainly atypical absences, tonic, and atonic seizures. Tonic seizures are the main feature of the syndrome, but they may not be easily recognized due to their frequent occurrence during sleep. Atypical absences may present in status and clinically as clouding of consciousness. In young children, arrest of psychomotor development is the rule, whereas intellectual impairment may be less pronounced in cases of later onset. Prognosis: the outcome is always poor because seizures are persistent for many years and all patients are cognitively impaired. However, in recent years many common features were recognized as shared by these two syndromes, and questions arose as to whether they are two distinct entities or subclasses of a single syndrome. Age of onset ranges from 3 to 8 years, and boys are more frequently affected than girls. The onset of aphasia is often insidious and progressive, with spontaneous improvements and aggravations during its course. The most common feature is verbal auditory agnosia, Myoclonic Status in Nonprogressive Encephalopathies this condition is considered for inclusion in the new classification of epileptic syndromes. The syndrome is characterized by the appearance of repeated atypical myoclonic status, combined with an impairment of attention in infants suffering from nonprogressive encephalopathies. Polygraphic records are useful to recognize more or less rhythmic asynchronous myoclonia that may not be evident clinically. Variable times may elapse between the loss of ability to understand language and the expressive aphasia. Neuropsychological and behavioral disturbances have been reported, but the most frequent findings are hyperkinesia and excitability. In fact, it is striking that children with such a severe handicap in understanding language only present only psychotic or autistic features when aphasia appears in early ages. The most common types of seizures are eyelid myoclonia, eye blinking, atypical absences, head drops and atonic fits in the upper limbs, automatisms, and occasionally partial motor seizures with secondary generalization. It has been stated that most cases have a unilateral primary epileptogenic region. Treatment with high-dose steroids was reported to yield the best results, and prolonged chronic or intermittent therapy may be necessary. Multiple subpial transection of the cortex to abolish epileptic discharges was used in a series of 14 children with acquired epileptic aphasia who had been unable to use language to communicate for at least 2 years: Sustained improvement was obtained in 11 of them. Classification of Diseases Frequently Associated with Epileptic Seizures or Syndromes the concepts behind the creation of this group were detailed in the introduction. Table 1 is quite comprehensive in relation to the wide range of items that may be considered within this framework. However, for simplicity, only the groups of diseases and the specific diseases that are frequently associated with epileptic syndromes are listed here (Table 3). Language is commonly affected, but the most striking symptoms are bizarre behaviors. In the majority of the symptomatic cases, language dysfunction and learning impairment never return to normal levels. Commission on Classification and Terminology of the International League against Epilepsy (1981) Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Commission on Classification and Terminology of the International League against Epilepsy (1989) Proposal for revised classification of epilepsies and epileptic syndromes. Epileptogenesis K Lukasiuk, Nencki Institute of Experimental Biology, Warsaw, Poland r 2014 Elsevier Inc. Introduction the term epileptogenesis describes the pathogenic process by which normal brain is transformed to epileptic brain, capable of generating spontaneous seizures. It is most often used in the context of acquired epilepsies and refers to the latency period from the brain damaging insult In such cases, an initiating event induces a complex cascade of changes in the brain leading to changes in structure and function that are morphological and physiological substrates of seizure activity. Epileptogenesis may also occur in genetic epilepsies and then it would refer to the time period preceding spontaneous seizures, when the brain becomes epileptic as a result of genetic programing. Additionally, it has been suggested that the same processes that underlie epileptogenesis triggered by epileptogenic insult can also continue to progress after the epilepsy diagnosis. The events initiated in the brain by epileptogenic insult leading to the decreased seizure threshold are poorly understood as yet. It is evident that the initiating insult initiates a dynamic sequence of events including numerous structural, cellular, and molecular changes happening over time, but it is not clear which of them are causative for epilepsy development. Although the risk factors for human epilepsy are known, there is no strategy to prevent epileptogenesis or reduce the chances of developing the disease in individuals at risk. Animals recover from status epilepticus and then, following a latency period of several days or weeks, start to express unprovoked seizures. Experimental animals develop hyperexcitability following insult and some of them develop also spontaneous seizures. Neurodegeneration has also been observed in other brain areas: amygdala and the surrounding entorhinal, perirhinal, and parahippocampal cortices, as well as in many extratemporal areas, including the thalamus. As shown in animal models of epilepsy, neuronal death is induced by insult but it can progress further during epileptogenesis and epilepsy. However, it has recently been shown that even substantial protection of hippocampal neurons does not prevent epileptogenesis and neurodegeneration is not needed for epileptogenesis in immature brain. In addition to neuronal cell death, axonal injury has been observed as a result of epileptogetic insult. Several animal models in vivo as well as in vitro preparations have been established and successfully used. Experimental models differ in terms of etiology, expression of spontaneous seizures, or presence and extent of brain pathology. It is produced by repeated applications of initially subconvulsive electrical stimulation of the selected brain area or application of a subthreshold dose of proconulsive compound, which following several kindling sessions starts to evoke convulsive seizures. Astrocytes participate in regulation of extracellular potassium, glutamate uptake, and synaptic glutamate concentrations, and produce and secrete various molecules including proteases or inflammatory mediators. Disturbances in astrocytic function 196 Encyclopedia of the Neurological Sciences, Volume 2 doi:10. Additionally, astrocytes can form a scar preventing growth of axons and blood vessels into the damaged brain area. One function of activated microglia is clearing dying cells from the damaged tissue. Activated microglia also release a number of compounds including inflammatory mediators, proteases, or trophic factors. Others can be neuroprotective (transforming growth factor b, brain-derived neurotrophic factor, neurotrophin-3, or nerve growth factor). Whether antiepileptogenic effect can be achieved by manipulation of astro- or microgliosis remains to be studied. Mechanisms involving immune and inflammatory mediators are emerging as an important player in epileptogesis. Therefore, inflammation and immune mechanisms are increasingly being studied and targeted in experimental antiepileptogenic therapies. Although a fraction of neurons generated in response to damaging stimuli eventually die, the remaining neurons integrate into the neuronal network but often display abnormal features. Additional black staining in panel B2 is derived from aberrant mossy fiber sprouting (red arrows). Animal studies indicate that sprouting is detectable already during epileptogenesis, before spontaneous seizures occur. This phenomenon has been proposed as one of the mechanisms of increased network excitability. However, recent experimental data indicate that decreasing sprouting does not prevent epilepsy development. Therefore, the functional role of axonal sprouting in epileptogenesis is still controversial. Loss of dendritic spines, changes in spine morphology, reduced dendritic branching, and abnormal growth of basal dendrites have been described following epileptogenic insults. Changes in dendritic morphology can result in impairment of neuronal connectivity. No effective strategy to stop epileptogenesis has been designed in animal models of epilepsy. However, some approaches have resulted in a favorable modification of the disease in terms of number of seizures or behavioral impairment. Pitkanen A, Bolkvadze T, and Immonen R (2011) Anti-epileptogenesis in rodent post-traumatic epilepsy models. Pitkanen A and Lukasiuk K (2011) Mechanisms of epileptogenesis and potential treatment targets. Channelopathy Brain-damaging insults induce both short- and long-lasting alterations in gene and protein expression and localization, as well as induction of numerous transcription factors and signaling pathways. One of the proposed molecular mechanisms of epileptogenesis is acquired channelopathy. Molecular analyses of ion channels after epileptogenic insults have revealed such changes, which can be directly associated with altered electrophysiological functions of neurons. Epigenetic Mechanisms Recent studies indicate that epigenetic mechanisms occur and possibly play a role in epileptogenesis. Antiepileptogenesis Several neurological conditions are associated with increased risk of epilepsy. This article is a revision of the previous edition article by Christopher G Goetz, volume 2, p 290, r 2003, Elsevier Inc. Paroxysmal or episodic ataxias are conditions that cause patients to fluctuate between severe incoordination, balance and gait disruption, and normal or near-normal function. In patients with onset in infancy or early childhood, there is usually mental retardation as one of the accompanying features. In children, an inborn metabolic disorder should be sought, such as urea cycle deficiencies, aminoacidurias, or disorders of pyruvate and lactate metabolism. In contrast, when episodic ataxia occurs in adults, several other considerations are important, including disorders related to drug ingestion, multiple sclerosis, intermittent hydrocephalus, and transient ischemic attacks. There are also familial forms of dominantly inherited periodic ataxia that appear to relate to defects in small ion channels, including those for potassium or calcium. The involved genes, which control neuronal voltage-gated potassium and calcium channels, are present throughout the nervous system, but predominantly in the cerebellum.
There are ample ways to take the risk and the fears away hiv infection early warning signs buy mebendazole 100 mg without a prescription, that contributing to the joys of motherhood hiv symptoms urinary tract infection cheap mebendazole 100 mg buy online. People with epilepsy need a comprehensive approach to diagnosis antiviral drugs for shingles order mebendazole 100 mg without prescription, treatment hiv infection rates in african countries discount mebendazole 100 mg buy line, rehabilitation hiv infection virus mebendazole 100 mg order mastercard, and counseling, and they may need additional help to overcome the negative consequences of their condition. The outcome of rehabilitation programs would be a better quality of life, improved general social functioning and better functioning in, for instance, performance at work, and improved social contacts. Stigma and Attitudes Stigma In spite of the significant clinical and therapeutic progress made in the past century, people with epilepsy still continue to suffer from stigma and discrimination, both in the developed and in the developing worlds. There is general agreement that stigma and exclusion are common features of epilepsy in both the developed and developing countries and a major contributor to the burden associated with the condition. Reducing the stigma of epilepsy is therefore key to reducing its impact and so improving quality of life. As a feature of many chronic health problems, stigma contributes to the hidden burden of illness. Health-related stigma is typically characterized by social disqualification of individuals and populations who are identified with particular health problems. High scores were correlated with worry, negative feelings about life, long-term health problems, injuries, and reported side effects of antiepileptic drugs. Attitudes Attitudes toward people with epilepsy are influenced by the degree of knowledge. After these people had been educated about epilepsy, these figures changed for the better. Quality of Life and Treatment Studies conducted in patients with refractory epilepsy suggest that the improvement of quality of life may be independent of the reduction of seizure frequency. Controlling the seizures does not automatically improve the quality of life because the other consequences of the condition often remain. Epilepsy must be given a higher priority by governments, policy makers, health care professionals, and communities, within all countries. Conclusion People with epilepsy are not only confronted with this health problem but also have to cope with a wide range of difficulties that affect almost every aspect of their lives. Many of these difficulties are a consequence of misconceptions, prejudice, and stigma. Even though epilepsy is one of the oldest disorders known, from some 4000 years ago until today, it was common belief, in many parts of the world, that people with epilepsy were seized by the gods or by the devil in some shape or form. Professionals need training in order to better understand these issues and in order to be able to offer people with epilepsy the services they deserve. People with epilepsy themselves need to know more about the condition in order to be able to make informed choices about their future. The general public needs to know the facts concerning epilepsy in order to change their attitudes toward people with epilepsy, thus creating social acceptance in society. Overall, the presence and frequency of seizures are the principal factors responsible for the decreased quality of life observed in epilepsy. This total cost includes the direct cost of the medical resources expended and indirect cost of decreased productivity. The indirect cost of epilepsy is particularly significant and valued at six times the direct cost. The role of surgery in the treatment of epilepsy is based on its safety and singular efficacy in clinical situations that are expensive, disabling, injurious, and sometimes fatal. Evaluation for Candidacy the comprehensive evaluation that leads to determination of epilepsy surgery candidacy is built on establishing the localization of the epileptogenic zone, that is, the region of brain tissue that is necessary and sufficient for the generation of seizures. As there is no test that specifically identifies the epileptogenic zone, the results of several structural and functional tests are compared with the objective of determining concordance of the results. The structural evaluation of the brain is key as partial seizures usually have an anatomical substrate. The functional evaluation serves to localize and characterize focal abnormalities, and also to assess the functional integrity of regions being considered for resection. Functional testing also provides a means to identify focal abnormalities that are independent of the structural abnormality, and therefore it is useful because the two types of abnormality may occur independently. It depicts anatomy with high resolution and identifies regions of structural abnormality that may indicate the epileptogenic zone. Video recording allows behavioral manifestation of the seizure which aids in the determination of the anatomic origin of the seizure. This is accomplished with either stereotactically placed depth electrodes or subdural grid electrodes. Behavioral testing immediately following the injection may indicate hemispheric dominance for language and the functional integrity of the memory system contralateral to the injection. How the diagnostic tests are brought together for the determination of surgical candidacy varies among institutions, and there is no consensus regarding the diagnostic requirements for epilepsy surgery. The weights placed on the individual tests within the evaluation are constantly evolving and therefore are not included in the algorithm. Candidacy Diagnoses Beyond localizing a suspected epileptogenic zone and assessing its functional status, the surgical evaluation serves to determine a more exact epilepsy syndrome diagnosis. The development of epilepsy surgery over decades has led to the identification of several surgically remediable syndromes. These syndromes have understood pathophysiologies and natural histories, are usually amenable to noninvasive evaluations, and have established surgical outcomes. Moreover, these syndromes predictably have a poor response to medical therapy and a good response to surgery. As epilepsy evaluation has become more sophisticated, the list of surgically remediable syndromes has grown. Acquired brain injury 164 Encyclopedia of the Neurological Sciences, Volume 2 doi:10. Although the diagnosis of a syndrome that has a poor natural history is one requirement for epilepsy surgery candidacy, there is no consensus as to when such individuals should be considered for epilepsy surgery. As the natural history of syndromes and their likelihoods of responding to medication become better understood, the time at which epilepsy surgery should be considered may be defined more clearly. Studies of the efficacy rates of successive antiepileptic medications have shown that the likelihood of efficacy decreases substantially if the first medication fails due to inefficacy. This has supported the general approach of not exhausting all medication options before considering epilepsy surgery. A more specific approach to the timing of epilepsy surgery may be based on the syndromic diagnosis and results of studies of seizure control with medical treatment for specific syndromes. Surgical Techniques A variety of operations may be performed in the surgical treatment of epilepsy. Deciding which operation is appropriate depends mostly on the epilepsy syndrome or localization. Cortical resection is the most commonly performed operation, and involves the resection of both structural and functional abnormality. These two types of abnormality often do not completely overlap, and the region generating seizures often includes a structural abnormality and the adjacent tissue that is structurally normal but epileptogenic because of its proximity to pathology. Alternatively, sometimes no structural abnormality is evident, and functional testing determines the limits of the resection. Various anterior temporal lobe resections are the most commonly performed epilepsy surgery procedure. A randomized, controlled trial of cortical resection against medical treatment has been performed. The other surgical procedures are performed when either cortical resection is not possible because of the colocalizing neurological function, or the epileptogenic region is either diffuse or poorly localized. Lesionectomy involves the resection of only the structurally abnormal tissue, and thereby minimizes the deficit because the pathological region lacks neurological function. It relies on the vertical columnar organization of function within normal cerebral cortex, and the propensity of epileptic discharges to spread horizontally across cortex. Instead, it disrupts cerebral cortex with cuts that are parallel to the columns, and thus function may be minimally affected and the spread of epileptic discharges impeded. Hemispherectomy and large multilobar resections are the most commonly performed procedures for diffuse unilateral hemispheric pathology that presents early in childhood. When performed early in life, cerebral plasticity allows the remaining hemisphere to assume much of the function of the resected hemisphere. Large resections, such as hemispherectomy, are rarely performed in older individuals because of the major neurological deficits that result from them. Drop attacks due to diffuse bilateral hemispheric pathology may be treated through a corpus callosotomy, a palliative procedure that disconnects the hemispheres and reduces seizure spread and severity. The stimulation occurs according to a cycle and with parameters that are adjustable through a radio frequency transmitter. Chronic stimulation of this nerve has been found to reduce seizure frequency, and may be effective when antiepileptic medications fail. J (1993) Update on surgical treatment of the epilepsies: Summary of the Second International Palm Desert Conference on the Surgical Treatment of the Epilepsies. This description contains all core elements of what was later called the focal dyscognitive seizures, including loss of consciousness. In some cases there is warning by what is known as the epigastric sensation, a systemic sensation which especially appertains to the digestive system. Epileptic Seizures and Syndromes of the Temporal Lobe A variety of conditions associated with focal seizures of presumed temporal lobe origin have been termed temporal lobe epilepsy. In this system, seizures of temporal lobe origin can be described as focal sensory seizures with elementary or with experiential sensory symptoms or as focal motor seizures with typical (temporal lobe) automatisms or with hyperkinetic automatisms. Furthermore, under the category continuous seizure type are listed the aura continua and limbic status epilepticus (psychomotor status). Under the axis epilepsy syndromes and related conditions, familial temporal lobe epilepsies and symptomatic (or probably symptomatic) focal epilepsies are listed. Familial temporal lobe epilepsy may be either mesial or lateral, and each is apparently due to different genetic etiologies. A key clinical distinction between the mesial and lateral forms is the common occurrence of auditory auras in familial lateral temporal lobe epilepsy. Problems occur because the term temporal lobe epilepsy has also been used to refer to conditions in which (i) the primary epileptogenic region is outside the temporal lobe but discharges preferentially propagate to the temporal lobe to produce typical seizures, (ii) an old and initially epileptogenic lesion leads to the development of an active temporal lobe focus in the sense of an independent secondary focus while the primary epileptogenic lesion has lost its epileptogenic properties (sometimes called secondary temporalization), and (iii) the epileptogenic region is in the lateral temporal lobe and gives rise only to neocortical-type seizures. Pure descriptive terms, such as psychomotor epilepsy, have obvious limitations because psychomotor seizures can originate outside the temporal lobes. The syndromes apparently differ in genetic and environmental factors, natural history, pathogenesis, and prognosis. A significant association between the severity of hippocampal sclerosis and younger age of onset of epilepsy but no definite relationship of hippocampal sclerosis to the duration of epilepsy have been found, implying that hippocampal sclerosis is not the consequence of long-standing seizures but that there is an agespecific sensitivity of the hippocampus to whatever causes hippocampal sclerosis. The prevalence of a positive family history of febrile seizures is higher in patients with late seizure recurrence and in patients with temporal lobe epilepsy treated surgically. It is assumed that the tendency for febrile seizures is genetically determined and inherited in a multifactorial way. Epidemiology Focal dyscognitive seizures (which has replaced the term complex partial seizures) are a common seizure manifestation, especially when anterior temporal lobe pathology is present. The term mesial temporal sclerosis has advantages because it better accounts for the fact that the amygdala, during examination, often shows equally severe pathology consisting of neuronal loss and gliosis to that of the hippocampus. The subicular complex, entorhinal cortex, and the temporal gyri are relatively resistant to cell loss. Hippocampal sclerosis is associated with other characteristic features, such as mossy fiber sprouting and selective loss of somatostatin and neuropeptide Y-containing neurons. The association between epilepsy and a sclerotic hippocampus was described as early as 1825, but at that time hippocampal sclerosis was believed to be an effect rather than a cause of epilepsy. Pathophysiology and Genetics the majority of focal epilepsies begin in early life. Hippocampal sclerosis is most commonly found in patients with a history of febrile seizures or status epilepticus in childhood, but it also has a lesser but significant correlation with birth trauma. A common denominator of many studies is that there might be a particular time frame (approximately 3 months to 5 years) during which noxious events are particularly harmful. Moreover, hippocampal sclerosis is often a bilateral condition, although frequently with a unilateral preponderance. The events that initiate the process of hippocampal sclerosis are unknown, but there is no doubt that the epileptogenicity of this disorder results from loss of specific neurons in the hippocampus and synaptic reorganization of surviving cellular elements with resulting hypersynchronization and hyperexcitability. Clinical Characteristics Typically, onset of seizures occurs at the end of the first decade. Dystonic posturing of one extremity can occur and is a valid lateralizing sign indicating contralateral ictal onset. Ictal automatisms frequently consist of oroalimentary symptoms, such as lip smacking and swallowing, and gestural Most ictal hallucinations are experiential phenomena in the sense of being recollections of past experiences. Psychic symptoms can be described as (i) intellectual and (ii) affective-emotional phenomena. Some aura symptoms may not have counterparts in normal human experience and cannot be described. Ictal autonomic phenomena can be classified as visceromotor symptoms and viscerosensitive sensations. Measurable autonomic visceromotor changes occur during the course of most temporal lobe seizures. When they characterize the clinical picture, the terms visceral or autonomic seizures are used. According to the effector systems, one can distinguish between cardiovascular, respiratory, pupillary, sudo- and pilo-motor, salivation, gastrointestinal, and genitourinary. Seizure Distribution Seizures typically occur randomly, with a common frequency of a few per month. There is no marked circadian preponderance of seizure occurrence, but drowsiness and light sleep (nonrapid eye movement sleep stage 1 facilitate seizures). Detailed neuropsychological examination, however, reveals various degrees of material-specific learning and memory deficits.
Bizarre Gait Bizarre gaits may be neurological or psychogenic (caused by psychiatric disturbances) in origin antiviral blu ray 100 mg mebendazole purchase free shipping. Astasia abasia (the inability to stand) is caused by midbrain antiviral resistance definition 100 mg mebendazole purchase fast delivery, thalamic hiv infection rate sri lanka purchase generic mebendazole from india, lenticular hiv infection using condom buy 100 mg mebendazole free shipping, and frontal lobe lesions or is due to psychogenic causes hiv infection who mebendazole 100 mg purchase with amex. They take a variety of forms, some suggesting recognized gait disorders such as hemiparesis and ataxia. However, often the gaits are bizarre and do not closely mimic common gait disorders. First, the signs are inconsistent with any neurological dysfunction and distraction with another task is often a method to bring out normal neurological function. The patient with a psychogenic gait disorder can often improve sufficiently to walk to the smoking area or candy machine. The third indication of psychogenicity is other psychogenic neurological signs such as give-way weakness or nonphysiological sensory losses. Many patients with psychogenic disorders are seemingly indifferent to their disability. Often, there is a history of other strange medical diagnoses suggesting psychogenic disorders. Treatment the goal of diagnosis, of course, is to identify underlying disease that is treatable so that normal walking can be restored. In general, the approach to gait disorders is to try to keep the patient active but safe. Unfortunately, gait disorders usually progress once present, and bone fractures such as hip fracture may be major consequences. Some patients may benefit from the use of a three-wheeled walker with hand brakes, and some may benefit from physical therapy. Physical therapy programs generally include therapeutic exercises, gait training, and psychosocial support. Rehabilitative efforts are often aimed at improving motor function, increasing range of motion, and building endurance. In addition, physical therapy is effective in building confidence and reasonable expectations, thereby reducing the fear of falling. Patients and their families should be instructed about safety measures, including correcting visual acuity, eliminating unnecessary drugs, improving lighting in the home, and instituting gait aids. While standing or walking, the person simply loses muscle tone and collapses or drops. History and additional workup for the following entities are important in uncovering and treating the causation of collapsing gait: See also: Ataxia. He was interested in science from an early age and synthesized commercially useful chemical compounds when he was still a schoolboy. He continued his laboratory research when he was a student at the University of Rochester and Harvard Medical School. After training in clinical pediatrics interspersed with stints of research at Columbia Presbyterian Medical Center and the California Institute of Technology, he went to the Walter Reed Army Medical Service Graduate School. There, he became interested in infectious diseases in developing countries, so he took a post as a visiting researcher in Melbourne, Australia. Gajdusek found that the Fore developed kuru from the ritualistic consumption of the brains of their newly dead ancestors. Positing the presence of an unusual infectious agent, he succeeded in transmitting the disease to chimpanzees by intracerebral inoculation of a brain suspension from affected patients. Gajdusek observed the similarity of kuru with the epidemic scrapie in sheep but was not able to isolate the slow viral agents. Some decades later Stanley Prusiner found that these agents are conformationally altered proteins, or prions, which could be either infectious or inherited. Through the years Gajdusek adopted several dozen children, usually boys, and brought them from southeast Asia to the United States. There he encouraged and supported them, especially in their educational endeavors. In the 1990s, the authorities accused him of child molestation based on the testimony of some of his adopted sons and also from frank entries in his notebooks. He spent his last years in Europe, apparently unrepentant about his confessed pedophilia. For over a half century, postnatal presymptomatic diagnosis through newborn screening has resulted in early detection and prompt treatment by lactose restriction. Because for infants the predominant source of galactose is lactose from milk and milk products, the early institution of a lactose-free diet in the neonatal period quickly resolves the neonatal manifestations of neonatal hepatitis, inanition, and sepsis. Response to dietary intervention has been generally satisfactory but not uniformly so. It is apparent that long-term neurological deficits and organ system dysfunctions seen in many treated patients cannot easily be explained by the direct or indirect toxic effects of high concentrations of metabolites, such as galactose-1-phosphate and galactose, accumulating proximal to the metabolic block. In sum, the etiology of toxicity in treated and untreated galactosemia is not fully understood. In some instances, infants have been noted to have diffuse cerebral edema and signs of increased intracranial pressure. After infancy, patients have been described with an encephalopathy characterized by choreoathetosis, hypotonia, tremor, and cerebellar signs. Some of these affected patients were treated late or even untreated, whereas others were reported as appropriately controlled by diet. A number of lactose-restricted patients have exhibited a distinct and slowly progressive neurological syndrome, with language delay and dysarthria, mental retardation, hypotonia, postural and action tremors, and ataxia. In these patients, brain atrophy and abnormal myelination were demonstrated on brain magnetic resonance imaging. It has become apparent that patients who are well treated with appropriate and early lactose restriction may show longterm complications and adverse sequelae, independent of delays in diagnosis and dietary compliance. These patients can present with primary or secondary amenorrhea and delayed development of secondary sex characteristics. Patients with treated galactosemia may variably exhibit growth delay, verbal dyspraxia, and other speech disorders. Cognitive deficits, poor school performance, and behavioral and psychological disturbances have also been reported in treated galactosemic patients. In one comprehensive survey, cognitive deficits in treated galactosemic children were independent of the age at which dietary restriction was begun (during the first half of infancy) and were unrelated to the mean galactose-1-phosphate level in the first and second years of life. Following a brief period of good health, infants often present in the neonatal period after the institution of feedings with breast milk or lactose-containing formulas. Manifestations of the disease in infants include anorexia, vomiting, failure to thrive, lethargy, hypotonia, jaundice, hepatomegaly, and cataracts. These infants show hypoglycemia, a particular susceptibility to Escherichia coli (and other Gram-negative organisms) sepsis, hyperchloremic acidosis, albuminuria, and aminoaciduria. If untreated, the clinical course can be one of the progressive deteriorations, with death from hepatic failure, inanition, or sepsis. Deficiency of the first step in the pathway, the phosphorylation of galactose by galactokinase, leads to galactose accumulation, synthesis of excess galactitol, and resultant cataract formation. The most frequent mutation in European populations, Q188R, results in almost complete loss of enzyme activity and, generally, a severe phenotype. This mutation has been associated with increased risk for premature ovarian insufficiency and verbal dyspraxia. In general, however, there is considerable variability and inconsistency in outcome among given mutations, and the etiologies of such variability, and of the chronic complications, are not understood. Enzyme assays in amniotic fluid cells, or detection of known family mutations, can be performed for prenatal diagnosis. Although this screen is independent of feeding history, the testing is not valid for patients who have had a recent blood transfusion. In such instances, further evaluation of the neonate may include parental enzyme analyses to detect heterozygosity, or molecular genetic analysis of the proband. In some centers, newborn screening may include measurements of free galactose, or of erythrocyte galactose-1-phosphate. Elevated galactose-1-phosphate levels occur in both transferase deficiency and epimerase deficiency. Confirmation of this diagnosis is made on electrophoretic mobility or molecular genetic analysis. Treatment Elimination of lactose-containing milk or formula will reverse the manifestations of the acute neonatal hepatitis presentation. Substitutes used for this purpose include soy-based formulas (which are lactose-free but contain low levels of galactose), or glucose-containing formulas made with casein hydrolysates. Elemental formulas containing no galactose are available, and have been used in early infancy to reduce accumulating galactose metabolites. Beyond infancy, dietary restriction of galactose can be complex, and includes avoidance of not only dairy foods and medications containing lactose, but also foods containing free and bound galactose. The latter include fruits, legumes, nuts, and vegetables containing galactose in complex carbohydrates, galactolipids, and galactoproteins. The monitoring of treatment is generally through measurements of levels of erythrocyte galactose-1-phosphate and urine galactitol. Importantly, these are secondary chemistries, reflecting the degree of lactose restriction. Given the variability of outcome and the chronicity of complications in treated galactosemia, such chemistries do not necessarily reflect a truly efficacious reduction of toxic metabolites. In this setting, counseling, guidance, and medical management of patients Diagnosis Transferase deficiency galactosemia should be considered in any infant with the neonatal hepatitis clinical syndrome as described in Clinical Features. In older children, galactosemia should be considered in patients with unexplained cataracts, cirrhosis, neurodevelopmental deficits, speech abnormalities, or movement disorders such as tremor or ataxia. If the clinical findings warrant consideration of the diagnosis, evaluation and testing should be performed even if the newborn screening test was normal. Definitive functional testing rests on erythrocyte transferase enzyme assay in a patient who has not had a recent blood transfusion. If the enzyme activity is abnormal, the enzyme defect can be further characterized by Galactosemia 391 and families must include not only prescriptive dietary restrictions, but also monitoring and intervention for the known array of chronic complications, including speech abnormalities, neurodevelopmental deficits, and hypergonadotropic hypogonadism. Waggoner D, Buist N, and Donnell G (1990) Long-term prognosis in galactosemia: Results of a survey of 350 cases. Petry K and Reichardt J (1998) the fundamental importance of human galactose metabolism: Lessons from genetic and biochemistry. New texts, previously considered lost or attributed to other authors, are still being discovered. He recognized and vigorously promulgated the notion that the brain, not the heart, is responsible for sensation and voluntary motion, these being the physical attributes of the actions of the rational soul. This supreme part of the Platonic tripartite soul, which Galen appropriated from Plato (c. Born in Pergamum (now Bergama in northwest Turkey), Galen enjoyed a privileged upbringing. Galen had hitherto studied philosophy, and thereafter approached medicine with a philosophical framework. He entered a long period of medical and philosophical training at Pergamum, Smyrna, and Alexandria from the most renowned teachers in the Roman Empire. In particular, he went further in meticulously describing all the ventricles of the brain, using the ox as his preferred subject. Galen made extensive use of animal vivisection, using pigs, goats, and some species of primates. Galen drew freely from that legacy, using it to consolidate his own work on animal subjects because human dissection and experimentation had ceased with the passing of Herophilus and Erasistratus and were never repeated in antiquity. His description of what occurs when a section of the spinal cord is severed is clinically impeccable: After the incision, in all the nerves which lie below the place where the transection has been made, both the two potentialities are lost, I mean the capacity of sensation and the capacity of movement, and also all the bodily parts of the animal in which they are distributed become insensitive and motionless, a result that is inevitable, clear and intelligible. Along with his ventricular experiments of living animals, Galen also attempted to catalog clinical cases of damage to the brain, with the condition of such patients being interpreted strictly in ventricular terms. Admittedly, his recording of such cases was not strictly systematic in a contemporary sense. Nevertheless, it showed an understanding of the importance of applying what was observed in animal experimentation to broadly comparable human cases. From the lungs, it then enters the left ventricle of the heart, where it is elaborated into vital pneuma. This transformation is made possible by the action of innate heat within the left ventricle, acting in concert with altered venous blood from the right ventricle. There is no contribution from a so-called natural pneuma made in the liver, and the concept of circulation of either pneuma or blood is misleading in Galenic physiology. Galen also allows the ventricles to elaborate psychic pneuma from the outside air, which was erroneously thought to enter the anterior ventricles through a communication with the olfactory bulb. Similarly, a purported communication between the optic tract and the anterior ventricles was thought to allow ventricular pneuma to enter the eye so that vision could occur. With the exception of the cranial nerves, which take their origin from the cerebrum, Galen stated 392 Encyclopedia of the Neurological Sciences, Volume 2 doi:10. Galen believed that the fourth ventricle allows psychic pneuma to pass into the alleged hollow center of the spinal cord and hence be distributed to all the nerves of the body. Gall became an expert anatomist and developed a technique of dissecting the brain by making horizontal cuts and tracing tracts from the base to the cortex. He surmised that the cortex was an extension of the white matter tracts that took their nourishment from the cortex. Gall considered the spinal cord as an anatomical entity, neither a chain of ganglia nor just a tail of the brain. He divided the human brain into 27 organs, each corresponding to a discrete human faculty, 19 of which were shared by other animal species. He suggested that speech came from the frontal lobe areas and sex was centered in the cerebellum. Gall left Vienna in 1805 when his doctrines and lectures were considered to be dangerous to religion by the government. In 1808, the French Academy critically reviewed his idea that skull shape mirrored brain development and considered it unscientific.
Rather hiv infection versus aids discount mebendazole 100 mg on-line, they can be explained by the existence of common pathogenic mechanisms in epilepsy and psychiatric disorders antiviral infection definition cheap 100 mg mebendazole amex. The Impact of Psychiatric Comorbidities in the Management of Seizure Disorders Prognosis for Seizure Control Patients with a history of psychiatric disorders preceding the onset of epilepsy appear to have a worse response to pharmacotherapy of the seizure disorder jurkat hiv infection cheap mebendazole 100 mg overnight delivery. For example rates of hiv infection are higher in __________ prisoners buy 100 mg mebendazole fast delivery, in one study of 780 consecutive patients with new onset epilepsy followed over a 20 month period hiv infection rates europe order 100 mg mebendazole, those with a psychiatric history preceding the onset of epilepsy were twofold more likely to develop treatment resistant epilepsy than those without. In a separate study of 138 consecutive patients with new-onset epilepsy, those with symptoms of depression and anxiety before the start of antiepileptic therapy were significantly less likely to be seizure free after 12 months of therapy. By the same token, several studies have now reported that patients with intractable temporal lobe epilepsy and a lifetime psychiatric history were less likely to achieve complete seizure freedom following an anterotemporal lobectomy. In addition, a history of depression in patients being considered for epilepsy surgery has been found to predict the development of recurrence of depressive episodes postsurgically, whereas the presence of postictal psychotic episodes have been associated with an increased risk of postsurgical mood disorders. This risk increased by 32-fold in the presence of a mood disorder and 12-fold in the presence of an anxiety or schizophreniform disorder. In fact, in patients with treatment-resistant epilepsy, the presence of comorbid depression or anxiety disorders account for a worse quality of life to a greater degree than the frequency and severity of the seizure disorder. Only occasionally have definitions been provided, although standards have been developed by the Commission on Epidemiology of the International League Against Epilepsy. Generally, in approximately one-third of newly identified cases, some classic antecedent is identified. The distribution of causes in Iceland is similar in most studies of developed countries. These are the causes usually considered by neurologists when discussing etiology with patients. Each is associated with a substantial increase in risk for epilepsy that persists for a long period of time. Cerebrovascular Disease Although the incidence of cerebrovascular disease is declining, more people are surviving the initial cerebrovascular insult. Thus, an increasing number of individuals are at risk for poststroke epilepsy, and the incidence of epilepsy in the elderly has increased largely due to these cases. This risk is increased for at least 20 years poststroke, although most develop epilepsy within 2 years of the insult. Many studies on the association of cerebrovascular disease and epilepsy fail to distinguish between acute symptomatic seizures and later epilepsy. Thus, it is difficult to identify clear risk factors for epilepsy as opposed to risk factors for early seizures. There is virtually no increase in risk of epilepsy for people with infarctions in the brainstem. Although data are inconsistent, there seems to be a low risk for epilepsy in people with lacunar infarctions. Embolic infarctions have classically been associated with early seizures, but this has not been consistently the case, and there is no clear association between embolic stroke and late seizures. There is a high risk for epilepsy in those with cortical infarction, and the risk may increase with increasing size of infarct. Because early seizures have their own risk factors, it may be that early seizures represent a surrogate for these factors, such as size or location of infarct. Risk for epilepsy is probably higher in people with intracerebral hemorrhage and subarachnoid hemorrhage. Neurological residual (probably a surrogate for size and location of lesion) and early seizures are the risk factors for epilepsy in these cases. There is a clear gradient with increasing risk for epilepsy with increasing severity of injury regardless of how it is measured. In those with severe injury, this risk remains increased for at least 15 years following injury. In addition to overall severity, factors associated with increased risk include metal fragments, intracerebral blood, and brain contusion. Furthermore, prevention of early seizures does not affect subsequent risk for epilepsy. Without early seizures, almost all cases develop epilepsy within 3 years of the infection, but the risk persists for 20 years or more for those with early seizures. The primary factor associated with higher risk is early or acute symptomatic seizures. In a small series of cases with brain abscess, one-third of patients developed epilepsy within 2 years of the infection. The occurrence of epilepsy has been found to be relatively constant throughout the course of the illness. There is no clear association with myoclonic symptoms, but the risk may be greater in black patients. Dementia is increasing in the elderly, and this also accounts for the increasing incidence of epilepsy in the elderly. Multiple Sclerosis Multiple sclerosis is typically considered a disease of white matter. The pathophysiology is not well understood for these phenomena, but it has been suggested that the occurrence is related to the presence of cortical junction lesions. Tumors Primary and metastatic brain tumors have characteristically been considered a cause of epilepsy. Certainly, in a number of patients with intractable epilepsy, tumors have been identified at the time of surgery, and it is not unusual to identify tumors in people with longstanding epilepsy. With current imaging techniques, the late identification of neoplasm should become less frequent. There seems to be a correlation between recurrent postoperative seizures and tumor recurrence. Approximately one-third of all cases of brain tumor in Western countries present with seizures, and the tumor is identified at the time of presentation. In an older epidemiological classification, these presenting cases were considered to be acute symptomatic seizures. However, a new recommendation by the Commission on Epidemiology of the International League Against Epilepsy classifies these seizures as unprovoked. Alcohol and Other Recreational Drugs Alcohol abuse has been consistently shown to be associated with an increased risk of epilepsy. Heroin is associated with a threefold increase in risk for epilepsy, and cocaine is associated with a nonsignificant twofold increase in risk for epilepsy. For these conditions, it is not clear that the usual definition of epilepsy is appropriate because the pathology changes throughout the course of the illness. Other Progressive Disorders Autoimmune diseases, such as lupus erythematosus, are associated with epilepsy, although there are no systematic studies of risk. The risk seems to be 6- to A number of factors that have been evaluated in epidemiological studies are associated with increased risk for epilepsy, but there is insufficient evidence to say that they are causal. In a population-based Icelandic control study, children with incident unprovoked seizure were 2. Socioeconomic Status Low socioeconomic status is associated with a higher incidence of epilepsy than high socioeconomic status. It is also associated with an increased risk for developing epilepsy in adults with unknown, genetic, or presumed genetic etiology. Thus, the relationship is not driven by the association between low socioeconomic status and known causes of epilepsy, such as traumatic brain injury and stroke. Children with febrile seizures have a sixfold increased risk for subsequent epilepsy. There is considerable debate about the mechanism of the association, but it is probably heterogeneous in some cases, lesional but present before the first febrile seizure, and in other cases genetic. Depression Studies suggest that a history of major depression is associated with a 2- to 19-fold increased risk for developing unprovoked seizures. Mesial Temporal Sclerosis Mesial temporal sclerosis is a frequent finding at surgery in people with intractable epilepsy of temporal lobe origin. This pathology probably occurs with increased frequency in people with epilepsy compared with the general population, but the finding in people with epilepsy is not specific for type or severity of epilepsy. Hypertension Suicidal behavior Suicidal behavior is associated with an increased risk for developing epilepsy. This is important because it may suggest that the three- to fivefold higher incidence of completed suicide in epilepsy may reflect a recurrence of premorbid suicidality. Hypertension seems to be associated with an increased risk for epilepsy independent of stroke. Hypertension may explain the bidirectional relationship between stroke and epilepsy. In the absence of stroke, left ventricular hypertrophy, a marker of severe hypertension, is associated with an increased risk for developing unprovoked seizures. It is important to note that diuretics, a first-line treatment for hypertension, are protective for the development of epilepsy. Stroke Risk Factors Genetics Genetics certainly plays a role in the development of epilepsy. Although there is little increased risk for epilepsy due to structural or metabolic causes, there is a threefold increased risk for first-degree relatives of people with epilepsy of unknown, genetic, or presumed genetic cause. This is not limited to people whose epilepsy used to be called idiopathic epilepsy, a category declared to be genetic. Other Factors Age (increasing risk with advancing age) and gender (increased risk in males) are the risk factors for developing epilepsy. Other epilepsy risk factors have emerged that are part of the neuropsychiatric spectrum of disorders that include epilepsy. Meanwhile, it is clear that additional research is necessary if we are to make further progress regarding prevention. Introduction Epilepsy presents a global problem affecting all ages, social classes, groups, and countries. Significant problems are often experienced by people with epilepsy in the areas of civil rights, education, employment, residential and community services, and provision of appropriate health care. It imposes enormous physical, psychological, social, and economic burdens on individuals, families, and countries. Yet in term of disability, for many patients it is these other factors, which determine whether they will or will not make a satisfactory life adjustment. The problems are universal but have the most serious impact in the developing world. The aforementioned social consequences are related not only to the severity of the condition but also stem from the concept of epilepsy held by the general public. These concepts and the prevailing prejudice may lead to rejection, denial of education, and isolation. Children and adolescents often suffer from overprotection within their families and at school. The stigma of epilepsy also has a great influence on the education of children and young people with epilepsy, and this quite often leads to isolation of these youngsters. In adult life, problems are reported with obtaining and retaining employment, which are confirmed by research findings. They may lose their functional independence, for instance by losing the ability to drive, which may then lead to social isolation. The social consequences of having seizures and of the treatment will be touched on, and finally the influence of the attitudes of society, stigma, and discrimination will be briefly discussed. The forced cry, the loss of consciousness, the fall, the twitching and the foaming at the mouth, they all suggest possession by the spirit. In Swaziland, most traditional healers mention sorcery as the cause of epilepsy, whereby an enemy sends spirits to invade the body and cause convulsions. In Senegal in some places, the beliefs are such that those who suffer from epilepsy are held in high esteem, which is totally different from the surrounding countries. In India, especially in the rural areas, epilepsy is often considered to be due to an evil spirit, which needs to be exorcised. This is done by tying the person to a tree, beating him, cutting a portion of his hair from the scalp, squeezing lemon and other juices on his head, or starving him, just to mention a few. Bystanders who witness a seizure will often spread water on the forehead of the victim or make him smell a leather shoe. In the so-called developed world, the misconceptions may be different, but they too lead to problems for people with epilepsy. In the Netherlands, seizures are known to have led to a judgment of unacceptable attention-seeking behavior that needs punishment. They have lower self-concepts than, for instance, children with asthma do, and they have more problems with peer relations. Obviously, the development of epilepsy and the unpredictability of the seizures have a profound influence on the quality of life of children and adolescents. Parents become overprotective and are afraid to grant the child autonomy between seizures. Furthermore, families having a child with epilepsy tend to bypass this child in common communication. Parents also express anxieties concerning the impact of a child with epilepsy on their other children. The social integration of young persons with epilepsy depends on their reactions to their condition and to their family and society. Parental disharmony and family dysfunction due to depression may lead to educational and social failure. Poor seizure control reduces the capacity to participate in social activities, resulting in greater isolation. Young people with epilepsy experience a real struggle in negotiating educational systems and work opportunities. The barriers that keep them back must be identified, in consultation with the medical profession, at an early stage. Each family has its own emotional response to seizures and epilepsy, and their doctor must listen to their feelings and experiences and provide suitable information. Leo Tolstoy said that all happy families resemble each other, but each unhappy family is unhappy in its own way.
Buy discount mebendazole 100 mg on line. Itchy ears – symptoms causes and other risk factors.
References
- Schwartz D, Amir L, Dichter R, et al. The use of a powered device for intraosseous drug and fl uid administration in a national EMS: a 4-year experience. J Trauma. 2008;64(3):650-654; discussion 654-655.
- Visentin S, Wu SN, Belardinelli L: Adenosine-induced changes in atrial action potential: Contribution of Ca and K currents, Am J Physiol 258:H1070-H1078, 1990.
- Gnekow AK, Falkenstein F, von Hornstein S, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for lowgrade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol 2012; 14(10):1265-1284.
- Sandoz, R. (1976). Some physical mechanisms and effects of spinal adjustments. Annals of the Swiss Chiropractic Association, 6, 91n141.
- Senz A, Nunnink L. Inotrope and vasopressor use in the emergency department. EMA 2009;21:342-51.
- Bella AJ, Sener A, Foell K, et al: Nonpalpable scarring of the penile septum as a cause of erectile dysfunction: an atypical form of Peyronieis disease, J Sex Med 4:226n230, 2007.