

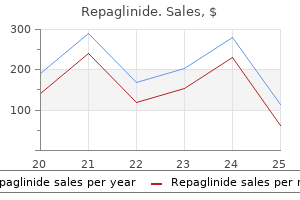
Repaglinide
| Contato
Página Inicial
Amber E. Proctor, PharmD
- Clinical Oncology Specialist, UNC Healthcare
- Clinical Assistant Professor, UNC Eshelman School of Pharmacy, Chapel Hill, North Carolina
https://pharmacy.unc.edu/news/directory/proctoae/
Early and sufficient blood product support should be given to those with massive blood loss and/or dilutional coagulopathy warning signs diabetes dogs cheap repaglinide 0.5 mg free shipping. Judicious use of fresh frozen plasma and platelets is required in patients with severe coagulopathy such as disseminated intravascular coagulation while the underlying condition is being treated diabetic alert dogs for sale cheap 1 mg repaglinide overnight delivery. Taking a history Assessing haemostatic capacity the main purpose of the history is to establish a clinical impression managing diabetes and shift work cheap repaglinide 0.5 mg mastercard, or profile diabetes diet education handout repaglinide 2 mg for sale, which indicates whether haemostatic capacity is normal or not and if there is a likely explanation for bleeding diabetic diet bananas discount repaglinide 0.5 mg otc. Rarely is it possible to absolutely exclude a disorder or make a positive diagnosis exclusively on the historical information. However, the history is critical, for example, there are some patients who definitely bleed after surgical challenges and yet the mechanism of their bleeding disorder cannot be characterized by laboratory investigations. Nevertheless, such patients should be considered to have a bleeding disorder and empirical treatment to reduce bleeding before surgical procedures should be planned. In contrast, patients with borderline abnormal laboratory tests but with no clinical bleeding tendency at times of haemostatic challenge should not be diagnosed as having a bleeding disorder. A comprehensive history is needed to assess the nature and extent of the bleeding, to guide clinical examination, and to logically prioritize investigations. An acquired bleeding tendency is much more common than a heritable genetic disorder and the most common cause of an acquired bleeding disorder is antithrombotic therapy, particularly oral anticoagulant therapy. Some patients who bleed abnormally during or after surgery have a mild underlying heritable haemostatic defect and so an important aspect of assessment is to determine if there is a heritable defect with late clinical presentation. Effective treatment depends on identifying the underlying cause of bleeding and anticipating when it is likely to be of clinical importance so that preventive therapy can be given at times of risk to prevent excess bleeding. Severity and pattern of bleeding the circumstances of the bleeding episode should be carefully assessed. Was the site of bleeding unusual, for example, a joint bleed in an adult with no previous history of abnormal bleeding Was there bleeding with previous haemostatic challenges such as surgery, trauma, dental extraction, and menstruation Bleeding symptoms over a long time period suggest a lifelong bleeding condition while recent-onset bleeding suggests an acquired disorder, although a mild lifelong condition can be unmasked at times of haemostatic stress such as surgery. Clinical assessment Presentation of a bleeding disorder varies from acute unexpected bleeding during or immediately after surgery to spontaneous unusual or excessive bruising, purpura, epistaxis, or other bleeding developing over several months. With increasing use of pharmacological thromboprophylaxis in both medical and surgical inpatients and increasing indications for long-term antithrombotic therapy, it is imperative to consider drug-induced bleeding during the initial evaluation of abnormal bleeding. Therefore, an important aspect of the assessment of a patient with an apparent acquired bleeding disorder is to determine if it is genuinely of recent onset. The major issues to be determined are as follows: · Is haemostatic capacity reduced or is there a nonhaematological cause for bleeding The extent of bleeding may be small (petechiae) or larger (bruising, also called ecchymoses). Bruising is common in patients with reduced haemostatic capacity but is also very common in patients who have no apparent defect of haemostasis. Very extensive bruising, particularly over soft areas that are not likely to be traumatized, and very large bruises in the absence of trauma (>5 cm diameter), are more likely in patients with reduced haemostatic capacity. Bruising over bony areas does not necessarily indicate an abnormality and many normal children frequently have several bruises over their knees and shins. When bruising is the result of a bleeding disorder, the pattern of bleeding may be suggestive of the type of underlying disorder, for example, ecchymoses suggesting coagulation factor deficiencies such as classical haemophilia or liver disease and petechiae suggesting thrombocytopenia or a vessel wall defect. Thrombocytopenia causes petechiae, typically when the platelet count is less than 20 × 109/litre. Petechiae may occur when there is platelet dysfunction or with vascular purpura, either of which can be congenital or acquired. In most patients, low-dose heparin does not appreciably increase surgical bleeding. However, it is important to review the drug charts from the time of the operation to ensure that the correct dose of heparin was given and that no other drugs that might cause bleeding were administered. Epistaxis Nosebleeds are common in children in the absence of a bleeding disorder. Bleeding in unusual sites Bleeding in an unusual site sometimes suggests a specific diagnosis. Gingival bleeding Gum bleeding in the absence of any other abnormal bleeding is usually due to gingivitis requiring improved dental hygiene. Family history of bleeding If a bleeding disorder has been diagnosed and characterized in another family member then the cause of bleeding may be easily identified. Menorrhagia Menorrhagia is a common problem and not specific for a bleeding disorder. Recent-onset menorrhagia in older women is likely to be due to a gynaecological cause. It is important to determine the pattern, for example, bleeding for several days with clots and particularly bleeding that interrupts normal lifestyle such as absence from work, is more likely to be abnormal and may be due to an underlying haemostatic defect. Very prolonged menorrhagia, rather than heavy bleeding, is more likely to be gynaecological. Drug history the most common cause of an acquired bleeding disorder is antithrombotic therapy. Increasingly, low-dose heparin for inpatient thromboprophylaxis and oral anticoagulant (warfarin, dabigatran, rivaroxaban, apixaban, edoxaban) and antiplatelet drugs (aspirin, clopidogrel, prasugrel, ticagrelor, cangrelor, nonsteroidal antiinflammatory drugs) in both inpatients and outpatients are responsible for bleeding. Dental extraction Bleeding after dental extraction is variable in the normal population. Bleeding lasting more than an hour, rebleeding, or late bleeding requiring suturing on more than one occasion suggests a bleeding disorder. Bleeding after extraction requiring blood transfusion even on one occasion suggests a bleeding disorder. Clinical examination Skin the skin should be inspected in its entirety for evidence of bleeding, noting the distribution (bony or soft areas), pattern (random or suggestive of nonaccidental injury), and size (petechiae or ecchymoses). Senile purpura occurs predominantly on the extensor surfaces of the hands and arms and the face. Purpura occur with amyloid, which may cause bleeding due to capillary fragility as a result of amyloid infiltration, or rarely an acquired deficiency of factor X. Amyloid may also cause Surgery It is important to ask specifically about all operations including circumcision and tonsillectomy. Abnormal bleeding during surgery or in the postoperative period may be due to antithrombotic therapy, such as warfarin or aspirin, that was not stopped. Abnormal surgical bleeding may be the first presentation of a mild or moderate heritable bleeding defect if there has been no previous haemostatic challenge, for example, a male having their first operation. Petechiae with a perifollicular distribution occur in scurvy, which may also present with more widespread bleeding into joints, gastrointestinal bleeding, and intracerebral bleeding. Other features include xerostomia, keratoconjunctivitis sicca, and hyperkeratosis. Purpura occurs with infections including meningococcal septicaemia and diphtheria, chickenpox, measles, and the haemorrhagic fevers of Ebola virus and Lassa fever. HenochSchönlein purpura is the most common allergic purpura and involves principally skin, joints, gastrointestinal tract, and kidneys. It typically occurs in children after an upper respiratory tract infection due to streptococcus. The rash consists of purpuric papules over the shins, thighs, and buttocks, sometimes with small ulcers, and the rash is associated with arthritis, nephritis, and abdominal pain. Mixed cryoglobulinaemia in patients with hepatitis C infection can produce extensive purpura in association with arthropathy and glomerulonephritis. Psychogenic purpura refers to unexplained bruising with preceding pain in association with anxiety. In patients with hereditary haemorrhagic telangiectasia, they occur predominantly in the skin of the hands and fingertips and are evident on the lips. Telangiectasias also occur in pregnancy and liver disease, usually on the face and chest. Rarely, large cavernous haemangiomas or aortic aneurysms can cause local consumption of coagulation factors and platelets resulting in a systemic bleeding disorder. Skin hyperelasticity, scars, papules, and plaques may indicate a collagen vascular disorder. EhlersDanlos syndrome, Marfan syndrome, pseudoxanthoma elasticum, and osteogenesis imperfecta are associated with a bleeding tendency due to abnormal platelet vessel wall collagen interaction. Unusual scars may be due to a dysfibrinogenaemia, EhlersDanlos syndrome, or pseudoxanthoma elasticum. Long-term steroid therapy and Cushing disease cause skin atrophy and bruising typically on the extensor surfaces of the hands and arms and on the thighs. Acute haemarthrosis presents as an acutely swollen painful joint resulting in joint immobilization. Repeated bleeds into a joint (target joint) produces chronic haemophilic arthropathy with features of both osteoarthritis (mechanical pain on movement) and rheumatoid arthritis (inflammatory pain at rest). Muscle haematomas occur in the iliopsoas, gluteal, calf, and forearm muscles and are more insidious than joint bleeds. Compartment syndrome can complicate large bleeds and fibrosis and contractures produce dysfunction and deformity. Large haematomas can cause pseudotumours, particularly when there is chronic rebleeding. These large, expanding soft tissue cysts produce mass effects including neuropathy and bone erosion and may produce chronic fistulas. Essential thrombocythaemia is a myeloproliferative disorder which is particularly associated with impaired platelet function but both bleeding and thrombosis occur. In patients with very high platelet counts, there can be increased consumption of von Willebrand protein causing an acquired von Willebrand syndrome. Patients with polycythaemia are particularly prone to chronic gastrointestinal bleeding. In these patients, bleeding may be due to thrombocytopenia, platelet dysfunction, coagulation factor deficiency, and production of dysfunctional factors. General aspects of examination In addition to identifying signs that may indicate the likelihood and type of a bleeding disorder, it is important to consider broader aspects. For example, is a patient anaemic due to iron deficiency as a consequence of chronic gastrointestinal blood loss Chronic liver disease, opportunistic infections, and other complications may be evident. Bleeding as a result of liver failure, renal failure, or paraproteinaemia should be considered. In children, the possibility of nonaccidental injury should be considered, particularly with multiple bruises around the head and neck, or a pattern of bruising in keeping with gripping or shaking. In a drowsy patient or a patient with raised intracranial pressure or an acute focal neurological deficit, there is the possibility of an intracranial bleed. Mucosa Telangiectasias are dilated small vessels that may be found in the skin and in the mucous membranes of the respiratory, gastrointestinal, and urinary tracts, vagina, eye, liver, and brain in patients with hereditary haemorrhagic telangiectasia (OslerWeberRendu syndrome). Coagulation tests are typically performed on plasma that has been separated from a venous blood sample by centrifugation. Thrombin generation takes place on phospholipid surfaces (provided by platelets normally) and so an artificial lipid preparation is added as the platelets are removed by the centrifugation. Most routine clotting tests use the time taken for a clot to appear as the endpoint of the assay. Blood is usually taken into tubes containing citrate, which chelates calcium and thereby prevents clotting. After centrifugation, the plasma isolated and recalcified during the clotting assay. Clotting tests are indicated in patients with a personal or family history of bleeding. They are not generally indicated as routine preoperative screening tests as they have very low sensitivity and specificity for surgical bleeding in unselected patients. Preoperative assessment of bleeding risk is better determined by identification of a personal or family bleeding history, which should then be investigated accordingly with specific tests including coagulation factor assays, von Willebrand protein level and function, and platelet count and function. Assessment of haemostasis the history is of primary importance in determining if haemostatic capacity is genuinely reduced. There is no absolute score that indicates an unequivocal diagnosis of a bleeding disorder but the higher the score, the more likely there is an underlying tendency to bleeding. Blood count and film examination Bleeding tendency increases with thrombocytopenia with a platelet count of less than 80 × 109/litre but severe bleeding rarely occurs in the absence of trauma with a platelet count greater than 20 to 30 × 109/litre. Pseudothrombocytopenia due to platelet clumping can lead to an erroneous diagnosis of true thrombocytopenia if the film is not examined for clumps. Conversely, a normal platelet count may be occasionally reported in patients with true thrombocytopenia, the normal count being an artefact, for example, due to the presence of a cryoglobulin. In these patients, the thrombocytopenia is readily apparent from examination of the blood film. Alternatively, a leukaemia or an acquired myeloproliferative disorder or myelodysplastic syndrome may be apparent from the blood count and film examination. Notes on the bleeding score: Mark the highest scoring box for each symptom (mark one box only for each symptom). The higher the score, the more likely there is an underlying tendency to bleeding. Therefore, it is on the basis of the history that a decision is made on the extent of laboratory testing. If these are normal but the history is suggestive of an underlying bleeding disorder, a more comprehensive laboratory assessment of haemostatic capacity is indicated. The cascade model of coagulation is no longer considered to represent the physiological process involved in coagulation. Heparin contamination is common on samples from hospitalized patients, even though the use of heparin flushes and sampling from catheters is often denied by clinical staff. In many cases it is necessary to obtain a venous sample from a fresh venepuncture.
These changes generally result in reduced red cell deformability leading ultimately to extravascular haemolysis diabetes scientific definition cheap repaglinide 0.5 mg without a prescription. These insults include infection diabetes medications chart 2015 1 mg repaglinide purchase mastercard, mechanical trauma diabetes test no food buy repaglinide with american express, and exposure to chemicals mayo clinic diabetes diet journal cheap repaglinide 0.5 mg buy online, heat diabetes type 1 webmd purchase repaglinide 0.5 mg overnight delivery, or venom. They often also cause intravascular haemolysis by directly lysing the red cell membrane. Infrequently, in utero infection of the fetus with Toxoplasma gondii resembles severe haemolytic disease of the newborn. Babesia microti, endemic in areas of the Northeast and Midwest of the United States of America, is transmitted by ticks and causes severe haemolysis during the erythrocytic phase of its life cycle. Bacterial infections, particularly Gram-negative organisms which produce endotoxin or proteolytic enzymes, may produce mechanical haemolysis by inducing disseminated intravascular haemolysis or red cell membrane damage via degradation of membrane phospholipids and proteins. Bartonella bacilliformis endemic to western South America causes Oroya fever characterized by fever, chills, musculoskeletal pain, and acute intravascular haemolysis. Chemical Drugs and chemicals known to cause haemolysis through direct oxidative damage are summarized in Tables 22. In most cases, the strong oxidant activity of these chemicals overwhelm normally functioning reduction mechanisms responsible for protecting haemoglobin and the red cell membrane. Variability in the absorption of the chemical or its metabolism Nonimmune acquired haemolytic anaemias Red cell survival may also be reduced by a number of noninherited, nonimmune mechanisms. As red cells circulate, they are vulnerable to a variety of insults that may cause structural or metabolic 22. Mechanical Mechanical fragmentation of erythrocytes can occur when foreign material is placed within the vasculature, when fibrin strands or platelet thrombi obstruct small blood vessels, or when direct physical forces compress superficial blood vessels. Thrombotic microangiopathies, which result in a microangiopathic haemolytic anaemia, are discussed in the following microangiopathic haemolytic anaemias section. Foreign material Mechanical haemolysis occurs most commonly with artificial valvular prostheses, particularly when accompanied by turbulent blood flow. Bacterial endocarditis and associated valvular vegetations can also cause fragmentation of red cells. Haemolysis also occurs in up to 10% of patients with transjugular intrahepatic portosystemic shunts. Increased cardiac output as a result of anaemia, exercise, or medications can increase the rate of red cell fragmentation. March haemoglobinuria Haemoglobinuria can occur in soldiers or joggers following extended periods of marching or running on a hard surface, or in karate or conga drummer enthusiasts following practice. This mechanical haemolysis appears to be the result of red cell compression in superficial blood vessels during the period of contact between the extremity and the hard surface. Treatment is unnecessary as the syndrome is otherwise symptomless and lacks significant clinical sequelae. Thermal haemolysis Normal red cells undergo fragmentation and lysis when heated to temperatures of 49°C or higher. The two most common clinical situations associated with heat-induced red cell lysis are the use of faulty blood warmers during transfusion or patients who have sustained extensive burns. Venom Haemolysis has been observed following bee and wasp stings, spider bites, and snake bites. The haemolysis occurs secondary to disseminated intravascular coagulation or as a result of proteolytic enzymes contained within the venom. The red cells of newborns do not have functional reduction mechanisms and thus are more sensitive to oxidant activity. Therapy consists mainly of supportive care, transfusion, control of blood pressure, and dialysis. Patients may present with fever, purpura, petechiae, anaemia, thrombocytopenia, and neurological abnormalities. The neurological sequelae include convulsions, coma, paralysis, delirium, and stroke. The peripheral smear demonstrates schistocytes, thrombocytopenia, and a reticulocytosis. Plasma exchange accomplishes one or more of the following: (1) removal of the antibody to the protease; (2) removal of large multimers of von Willebrand factor; and/or (3) replenishment of normal protease. In patients who do not initially respond to plasma exchange, a thorough re-evaluation for alternative diseases should be undertaken. The addition of rituximab should be considered, although the optimal dose and schedule have not been established. Cryo-poor supernatant contains markedly reduced levels of normal von Willebrand factor which is believed to enhance the formation of microthrombi in some patients. Additional reported therapies in refractory or relapsing patients have included Cytoxan, ciclosporin, vincristine, bortezomib, mycophenolate mofetil, Nacetylcysteine, and splenectomy. This patient had recurrent thrombocytopenic purpura and the marked fragmentation of the red cells together with microspherocytosis is evident on the blood film. In thrombotic microangiopathies, the red cells are fragmented during their forced passage through vessels containing microthrombi. Shiga toxin-mediated haemolytic uraemic syndrome Shiga toxin-mediated haemolytic uraemic syndrome is primarily, but not exclusively, a disease of childhood. The disorder consists of widespread damage to the vascular endothelium and fibrin deposition. These pathological changes are frequently most severe in the renal arterioles and glomerular capillaries. Numerous reports have documented the development of haemolytic uraemic syndrome following infections with toxin-secreting strains of Escherichia coli (strain O157:H7) or shigella. Initial nausea, vomiting, and diarrhoea can develop into severe abdominal pain and bloody diarrhoea. Acutely, the child may develop hypertension, oliguria, purpura, bleeding, and anaemia. Plasma exchange may be useful in some patients, and immunosuppressive therapy should also be considered in patients with antibody-mediated disease. Evidence-based focused review of the treatment of idiopathic warm immune haemolytic anemia in adults. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. Donor-derived red blood cell antibodies and immune hemolysis after allogeneic bone marrow transplantation. Prophylactic antigen-matched donor blood for patients with warm autoantibodies: an algorithm for transfusion management. Antibodies to von Willebrand factorcleaving protease in acute thrombotic thrombocytopenic purpura. Other elements-these include (1) extracellular matrix-promotes platelet adhesion, cellular migration, cell proliferation, and endothelial and smooth muscle cell interactions; (2) smooth muscle cells; and (3) adventitia. They adhere to damaged vessels where subendothelial matrix is exposed, aggregating to form an initial plug that prevents blood loss by occluding the breach in the vessel wall. These processes are very complex and involve highly controlled pathways of interaction between cells, glycans, and membrane-bound and soluble proteins of coagulation and fibrinolysis, as well as their cognate inhibitors. Thrombosis-this is an abnormal state leading to formation of a clot obstructing blood vessel flow; dislodgement leads to thromboembolism. Inhibitors of the coagulation reactions-there are numerous inhibitors of the reactions involved in blood coagulation, which are essential for the temporal control and safety of the process. The haemostatic plug consists of a mass of platelets, red blood cells, and leucocytes enmeshed in interlocking strands of insoluble and cross-linked fibrin fibres that plug the leak. Once formed, the haemostatic plug is gradually replaced by new tissue as a part of wound healing. This process requires lysis of the blood clot by the fibrinolytic system and subsequent ingrowth of new cells. Thus, haemostasis is not an isolated phenomenon, but is one component of the defence mechanisms that lead to eventual wound healing. Thrombosis, as opposed to haemostasis, is a pathological state in which a clot is formed that partially or completely obstructs the flow of blood within the blood vessel and sometimes dislodges to become an embolus. To understand the biology of haemostasis and thrombosis, it is necessary to know the roles of the vessel wall, the platelets, the coagulation and fibrinolytic systems, and their respective inhibitors. All blood vessels are lined by an intima consisting of a monolayer of endothelial cells that rest upon a loose network of tissue called the extracellular matrix. In addition to the intima, larger and intermediate sized arteries contain two other layers: the media, composed mostly of smooth muscle cells, and the adventitia, consisting largely of connective tissue, nerves, and nutrient vessels. These three layers also exist in veins, but the media and adventitia are much less distinct and are not visible in the smaller arterioles, venules, and capillaries. This system, so delicately controlled and normally maintained in a dynamic equilibrium, is strongly influenced by components involved in inflammatory and other defence mechanisms in the host. An integrated understanding of these processes offers the potential for improved means to predict the adverse complications of many diseases and ultimately to prevent their occurrence. Endothelium Internal elastic lamina Intima Media Introduction Fluid blood is contained within the vascular tree, but as a result of minor trauma that occurs during the wear and tear of everyday living, leaks occur in the blood vessel wall that must be sealed by a solid impermeable fibrin clot in order to prevent significant blood loss. The clot is formed from clotting factors in flowing blood and is located and restricted to the site of the leak without dissemination throughout the vascular tree. The intima consists of a layer of endothelium that is exposed to the circulating blood. The subendothelial matrix lies below the endothelium and is separated from the media by the internal elastic membrane. These early blood vessels expand into a vascular tree under the influence of two major hormones, angiopoietin 1 and 2, that bind to a family of tyrosine kinase receptors called tie-1 and tie-2 (tyrosine kinase plus Ig and epidermal growth factor-like domains) on endothelial cells. Endothelial cell structure is also dependent upon the integrin family of molecules and interactions with the extracellular matrix. Endothelial cells are heterogeneous in appearance, function, and genetic regulation. In the brain, endothelial cells form very tight junctions with one another to preserve the bloodbrain barrier; in the spleen and liver, the interendothelial gaps are wide, permitting soluble and cellular trafficking between blood and the extravascular space. The microenvironment also plays an important role in regulating endothelial cell function. Haemodynamic forces, including hydrostatic pressure, and shear stresses and strains can influence endothelial cell structure and function. Endothelial cells contribute to haemostasis by their contributions to vascular tone and procoagulant, anticoagulant, fibrinolytic, and antifibrinolytic activities. Vascular tone Vasoregulatory substances produced by endothelial cells are shown in Table 22. On the other hand, the most important vasoconstrictors are endothelin and angiotensin 2. Anticoagulant properties the anticoagulant properties of the endothelial cells are shown in Table 22. Prostacyclin not only causes vasodilation, but it is a potent inhibitor of platelet aggregation. An important anticoagulant function of endothelial cells is the expression of thrombomodulin, a transmembrane-bound protein that acts as a receptor for thrombin. Endothelial cells contribute to the control of coagulation by synthesizing tissue factor pathway inhibitor, which inhibits the tissue factor-mediated initiation of the clotting reactions. They also synthesize glycosaminoglycans such as heparan sulphate and other proteoglycans that inhibit thrombin via their interaction with antithrombin. Endothelial cells also secrete fibrinolytic factors including prostacyclin and tissue plasminogen activator, among others. It is released into the circulation in multimers of heterogeneous molecular mass ranging from 1000kDa to about 20 000kDa. Receptors the receptor function of endothelial cells plays an important role in haemostasis and thrombosis (Table 22. Thrombin cleaves the C-terminal end of the receptor, which then binds to the remaining cell-associated protein (a so-called tethered ligand) and triggers intracellular signalling through G proteins, resulting in activation of endothelial cells. Urokinase plasminogen activator receptors are not found on resting endothelial cells, but are found on those involved in angiogenesis. There are a number of adhesive receptors on the surface of endothelial cells, as shown in Table 22. P-selectin is rapidly internalized by the endothelial cell, but this is followed by expression of another cytoadhesive molecule, E-selectin, which is necessary for continued adherence and rolling of neutrophils along the endothelial cell surface. Extracellular matrix the extracellular matrix is a complex, heterogeneous structure beneath the endothelium with a number of constituents that contribute to haemostasis and thrombosis. The matrix proteins promote platelet adhesion, cellular migration, cell proliferation, and endothelial and smooth muscle cell interactions. The collagens are synthesized by endothelial cells, smooth muscle cells, and adventitial fibroblasts. The proteoglycans constitute a heterogeneous group of molecules composed of a core protein attached to a glycosaminoglycan. The precise role of all of the proteoglycans is not known, but some attach to collagen and are necessary for maintaining the structure of the vessel wall. The matrix also contains elastin, which is secreted by endothelial and smooth muscle cells as tropoelastin that is converted to mature elastin in the matrix where it is assembled into fibres. One function of elastin is simply to maintain the elastic structure of the vessel wall. This substance is found interspersed between smooth muscle cells as well as the matrix. It may also function in cell migration from the vessel wall to the extravascular space. Fibronectin, vitronectin, and laminins are also components of the extracellular matrix which function in fibrinolysis and platelet adhesion. They are secreted as proenzymes and converted to active enzymes that require zinc or calcium as cofactors. Their activities in matrix degradation and repair are controlled by tissue inhibitors of metalloproteinases.
As can be seen diabetes symptoms pregnant trusted 2 mg repaglinide, when viewed in this manner diabetes definition of who best order for repaglinide, the clotting reactions appear as a waterfall or cascade type 1 diabetes definition who generic 1 mg repaglinide mastercard, hence the terms waterfall or cascade hypotheses diabetes in dogs merck buy generic repaglinide pills. It was also pointed out that defects in the intrinsic system could lead to haemorrhage in affected patients even though the extrinsic system was intact and vice versa diabetes prevention questionnaire purchase repaglinide american express. Further work demonstrated that the clotting reactions leading to a haemostatic plug were controlled in large part by cell surfaces which modulated the reactions. It has a molecular mass of about 34 to 40kDa and serves to inhibit the initiation of coagulation. The factor Xa then associates with its cofactor, factor Va, on the platelet surface to rapidly convert prothrombin to large amounts of thrombin sufficient to clot fibrinogen. Fragment 1+2 derived from the N-terminal portion of prothrombin can also be detected after thrombin is formed. Activation peptides from several of the coagulation factors as well as complexes of activated factors with their inhibitors can also be found in the circulation. These observations strongly suggest that small leaks in blood vessels that occur during the stress and strain of everyday living are repaired by the ongoing formation of haemostatic fibrin clots. The fibrin plug is then removed by the fibrinolytic system following the formation of new tissue. This factor Xa in the presence of its cofactor Va then converts large amounts of prothrombin to thrombin sufficient to clot fibrinogen. The mass of aggregated platelets upon which these reactions take place is localized to the damaged area of the vessel wall. Role of the endothelial cells, vessel wall, and inhibitors the mass of platelets interspersed with fibrin forms a plug at the site of a leak in the vessel wall where the endothelial cell monolayer is disrupted. The question arises as how the haemostatic plug is confined to the damaged area of the vessel wall. In this way the fibrin clot sealing a leak in a blood vessel wall is confined precisely to that site such that extension of the clot does not occur under normal circumstances. The active enzyme in the fibrinolytic system is plasmin, which is derived from its precursor, plasminogen. Another activator of plasminogen in vivo is single-chain urokinase, but this appears to be more important for degradation of matrix proteins. Thus, clots can be lysed without interference from inhibitors, yet free plasmin in the circulation will be rapidly inhibited by its inhibitor. The final protolytic fragments resulting from plasmin degradation of fibrin are two molecules of fragment D and one of E. Plasminogen Plasminogen is synthesized in the liver and has a molecular mass of about 92kDa. It is composed of a single chain and exists in two forms in the circulation: Glu-plasminogen and Lys-plasminogen. Gluplasminogen has an N-terminal glutamic acid and is larger than Lysplasminogen, which is formed in the circulation by plasmin cleavage of an arginyl bond at position 78 of the Glu form, leaving lysine as the N-terminal residue. Thus, Lys-plasminogen is in close proximity to fibrin and protected from the action of antiplasmin. When activated, plasminogen is converted to active two-chain plasmin with a serine-active site on the heavy chain that is connected to the light chain by disulphide bonds. It is still clottable by thrombin, although much more slowly than native fibrinogen. Fragment X gives rise to fragments Y and D, and fragment Y is further proteolysed to give rise to a second fragment D plus fragment E. These fragments can be detected in a simple laboratory test using antifibrinogen antibodies coated to latex particles. However, the test is nonspecific and does not distinguish between the fibrinogen or fibrin degradation products which are quite similar, since the only difference between fibrin and fibrinogen is the absence of the small fibrinopeptides A and B in fibrin. Elevated levels of this inhibitor have been associated with arterial and venous thromboses. Its main function is in wound healing and vasculogenesis, and it is active in proteolysis of the extracellular 22. By binding to fibrin, antiplasmin competitively inhibits the binding of plasminogen to fibrin. However, when plasminogen within the fibrin clot is converted to plasmin, the latter is protected from inhibition by antiplasmin. Under normal conditions, fibrin clot formation is always accompanied by fibrinolysis. Although much is still unknown, it is generally accepted that both the coagulation and fibrinolytic systems are related to the general process of inflammation involving several other host defence mechanisms. Molecular biology and biochemistry of the coagulation factors and pathways of haemostasis. Long- standing bleeding symptoms suggest a lifelong condition, whereas recent-onset bleeding suggests an acquired disorder. If a bleeding disorder has been diagnosed and characterized in another family member, then the cause of bleeding may be easily identified, but the absence of a family history does not exclude a heritable disorder. Aside from general supportive care, specific therapy can be given when a defined haemostatic abnormality is identified. The intrinsic pathway of coagulation: a target for treating thromboembolic disease Vitamin K should also be given to critically ill patients and those with liver disease. Platelet function analysis Platelet function can be assessed at high and low shear. The test is performed on a citrated blood sample within 4 h of sample collection and is abnormal in the presence of low von Willebrand protein activity or platelet function defects. Thrombocytopenia causes prolonged closure and so the test requires a normal platelet count in order to assess platelet function. Aggregation studies are performed typically on platelet rich plasma prepared by slow centrifugation of citrated blood within 4 h of sample collection. There is a poor correlation with bleeding tendency except in specific congenital disorders characterized by severe platelet dysfunction, for example, Glanzmann thrombasthenia and BernardSoulier syndrome. Bleeding time the bleeding time is used much less frequently now that platelet function analysis at high shear is readily available. The bleeding time does not predict surgical bleeding and is largely no longer considered a test with clinical utility. These techniques include thromboelastography, thrombin generation, and tests of fibrinolysis. Interventional radiology with arterial embolization can be used to stop localized bleeding, for example, from the gastrointestinal tract. Specific issues Drug-induced bleeding the most common cause of an acquired bleeding disorder is anticoagulant therapy. Approximately 1 in 100 of the United Kingdom population are now receiving long-term oral anticoagulant therapy. A dilute plasma thrombin time such as the Hemoclot assay is useful for thrombin inhibitors (dabigatran) and drug-specific calibrated anti-Xa assays are required for factor Xa-inhibitors (rivaroxaban, apixaban, edoxaban). A specific antidote to dabigatran (idarucizumab, a monoclonal antibody fragment that binds to dabigatran) is licensed, and factor Xainhibitors. Antiplatelet drugs Platelets are integral to thrombin generation and antiplatelet drugs can be considered as anticoagulants, hence their ability to prevent thrombosis. The pharmacological half-life of antiplatelet drugs is determined by the mechanism of inhibition of platelet function. The lifespan of a platelet is typically 10 days and so 10% of the platelet pool is replaced each day. Once there is 50% replenishment (5 days after stopping therapy), platelet-dependent haemostatic capacity is usually adequate for normal blood coagulation. It is essential to examine the drug and infusion charts and check that the dose of any drug that may affect haemostasis is not excessive. It is also imperative to determine if the site of surgery is the only site of bleeding. If this is the case, for example, there is no bleeding from venepuncture sites or an endotracheal tube, and there is no history of previous abnormal bleeding, then depending on the results of coagulation tests it is important to keep the possibility of anatomical surgical bleeding as a likely possibility. In some cases of severe bleeding the patient may have to return to theatre to look for a bleeding point. Critically ill patients There are many potential acquired disorders of haemostasis in critically ill patients. A coagulopathy due to vitamin K deficiency occurs within a few days in critically ill patients with no oral intake. Parenteral vitamin K supplementation should be used routinely to prevent bleeding in the critical care setting. Standardized protocols for the early administration of blood products in patients receiving massive blood transfusion are increasingly used to ensure adequate replacement of clotting factors and platelets in order to prevent or at least limit the development of coagulopathy. In addition, abnormal bleeding is not usually apparent until the platelet count is below 80 × 109/litre, when platelet function is normal. Gestational thrombocytopenia occurs in 5% of pregnancies but the platelet count is rarely less than 80 × 109/litre and it is not associated with an increased bleeding tendency. Renal disease Bleeding risk increases with the degree of renal impairment and is due to a defect of plateletvessel wall interaction as well as impaired platelet function. Platelet function tests may give variable results that do not correlate with bleeding risk. Anaemia contributes to the bleeding tendency due to a reduction in platelet vessel wall contact as the haematocrit decreases. Bleeding is most commonly into the skin and mucous membranes and gastrointestinal haemorrhage is common. Haemodialysis and peritoneal dialysis reduce the bleeding tendency without any appreciable effect on tests of platelet function. Correction of anaemia by transfusion or erythropoietin therapy to maintain the haemoglobin above 100 g/litre is beneficial. Administration of conjugated oestrogens has also been reported to reduce the bleeding tendency. Regardless, the level of von Willebrand protein appears to be an important continuous variable influencing the coagulation phenotype. The first apparent manifestation of this may be excessive surgical bleeding and as a result the patient is considered to have an acquired bleeding disorder. It can be difficult to establish a diagnosis of a mild reduction in von Willebrand protein in the immediate postoperative period as levels rise due to the stress response. Consequently, it is prudent to re-evaluate patients several weeks after an episode of abnormal surgical bleeding. The liver is the site of synthesis of the majority of proteins involved in haemostasis. In cirrhosis, there is deficient production of coagulation factors compounded by production of dysfunctional factors (due to defective post-translational carboxylation) with thrombocytopenia due to portal hypertension with hypersplenism and in some case defective marrow platelet production. In obstructive jaundice, there is production of dysfunctional factors which respond initially to intravenous vitamin K. In advanced liver disease and hepatoma, there may be additional dysfibrinogenaemia. Hypofibrinogenaemia with a fibrinogen level less than 1 g/litre occurs with fulminant hepatic failure. Reduced clearance of tissue plasminogen activator may cause hyperfibrinolysis which contributes to bleeding. Parenteral vitamin K should always be considered and frequently a trial of therapy is required, for example, 10 mg daily for 3 days. Thrombocytopenia Many drugs result in a reversible idiosyncratic thrombocytopenia. Notable exceptions are quinine and gold-induced thrombocytopenia which are severe. An evaluation of drug history and cessation of possibly implicated drugs is essential in patients with acquired bleeding who are found to be thrombocytopenic. Other commonly used drugs for which there is good evidence for drug-induced thrombocytopenia include amiodarone, atorvastatin, carbamazepine, cimetidine, diclofenac, digoxin, ranitidine, co-trimoxazole, and vancomycin. Cytotoxic drugs produce a dose-dependent suppression of bone marrow platelet production and thrombocytopenic bleeding is common in oncology practice. A family history of thrombocytopenia or a lifelong history of a relatively stable, though low, platelet count is suggestive. Therefore, abnormal bleeding should not necessarily be attributed to a mild reduction in platelet count. Over the age of 65 years the lower limit of normal is around 120 × 109/litre and Acquired haemophilia and acquired von Willebrand syndrome Acquired inhibitors are rare and most often autoantibodies. Hyperfibrinolysis A bleeding tendency due to heritable defects of fibrinolysis is exceptionally rare. A single episode of abnormal surgical bleeding may not be readily explained but this should be taken into consideration at times of future surgery so that mechanical rather than pharmacological thromboprophylaxis is used and any antiplatelet or anticoagulant therapy stopped with certainty. Guidelines for the laboratory investigation of heritable disorders of platelet function. Bleeding in the neonate Bleeding in the neonate may be due to a rare heritable defect or an acquired abnormality occurring in utero or soon after delivery. Management of bleeding Acute bleeding Effective treatment depends on a critical assessment of the extent and nature of bleeding and the likely cause. When a defined haemostatic abnormality is identified, specific therapy can be given. Nonhaematological causes of bleeding should be managed appropriately, for example, dialysis and red cell transfusion in patients with renal failure. Vitamin K should be given to critically ill patients and patients with liver disease. Early and sufficient blood product support should be given to patients with massive blood loss and to those with dilutional coagulopathy.
As the cognitive deficits progress there is worsening of language function and semantic memory diabetes treatment kidney disease discount repaglinide express, and behavioural problems can be prominent diabetes prevention teaching plan purchase cheap repaglinide line. Agitation diabetes vitamins supplements order repaglinide 1 mg without a prescription, restlessness diabetes diet create your healthy-eating plan repaglinide 1 mg lowest price, wandering diabetes type 1 how long can you live cheap repaglinide 0.5 mg, and disinhibition can cause a considerable burden for carers. The final stages of the disease are characterized by reduced speech output (or mutism), ambulatory difficulties, dependence, and incontinence. There is considerable variation in the time from presentation to death: individuals diagnosed in their late 60s to early 70s have a median lifespan of 710 years, but this is reduced to three years when diagnosis is made in the 90s. In the final stages, there can be greatly increased rigidity and joint contractures. Mutations result in increased production of abnormal forms of A Impaired clearance of abnormal A forms results in accumulation Increase in A oligomers Deposition into A plaques Inflammatory responses ( A deposition, inflammation, and oxidative stress leads to tau pathology with tangle formation and atrophy; these downstream events lead to and correlate with neuronal dysfunction and cognitive symptoms. Neurological examination is unremarkable in the early stages, although increased tone (often frontal resistance, or gegenhalten, in type) and mild extrapyramidal features can occur as the disease progresses. Neuropsychological assessment characteristically shows early impairment in delayed verbal recall of new material, followed by reduced category fluency (in which individuals are asked to generate exemplars from a given category. Genetic testing for mutations in the causative genes may be appropriate in patients with young onset disease, particularly where there is a family history. Advances in genetic technology allow for several genes to be screened concurrently. Genetic testing should only be done with specific consent and following appropriate counselling, due to the implications for other family members. In contrast to testing for causative mutations, assessing genetic risk factors. In clinical trials, the cholinesterase inhibitors consistently achieve modest improvements in cognitive function compared to placebo. There is no evidence that they alter the overall course of the disease, although withdrawal of treatment in moderate-severe disease has been shown to worsen cognition and function and to increase the risk of nursing home placement. Memantine has usually been used in patients not tolerating cholinesterase inhibitors, or those with more advanced disease. There is some evidence that combined treatment with a cholinesterase inhibitor and memantine may provide benefit in some patients. There is less good evidence for how best to treat other aspects of the disease including depression, agitation, and psychotic phenomena; in such cases, input from a specialist psychiatrist is recommended. Environmental modification and appropriate nursing input should be considered prior to instigation of medication. Atypical antipsychotic drugs can be useful for treating severe neuropsychiatric symptoms, and risperidone is licensed for the short-term treatment of aggressive behaviour. However, these drugs are associated with an increased long-term risk of mortality and stroke in demented individuals, and so should be used cautiously and only when the benefits outweigh the risks. Several other approaches, targeting different aspects of the amyloid cascade, neuro-inflammation, and tau pathology, are in various stages of development/implementation. It is crucial to provide medical and psychological support to patients as well as to their families and carers. During the progression of the disease there will be different goals at different stages, ranging from aiding failing cognitive function in the setting of independent living, to managing behavioural problems and aggression, and eventually to providing full supportive nursing care. There is great variation in the rate of progression, but depending on the age and stage of disease at diagnosis, on average, patients spend several years in the mild or minimal stages (although it can be as long as 510 years), between 4 and 5 years in the moderate disease stages, and, depending on the quality of care in the dependent stages, a year or more requiring full nursing care. Nonpharmacological treatment the mainstay of treatment is social support and increasing assistance with day-to-day activities. Issues such as establishing fitness to drive, and financial planning while the patient has capacity to do so, are important and should be discussed early in the course of the disease. Arnold Pick (18511924) first described patients with both progressive aphasia and associated severe left temporal cortical atrophy postmortem, and patients with behavioural disturbances associated with frontal lobe atrophy. In 1910, Alzheimer described the histological changes in patients with focal lobar degeneration as distinct from the syndrome that bears his name, describing both argyrophilic intracytoplasmic inclusions (Pick bodies) and diffusely staining ballooned neurons (Pick cells). More recently it has become clear that the spectrum of pathology that accompanies the clinical syndromes within the frontotemporal dementia spectrum is much broader, with a range of distinct inclusions as described next. A significant proportion of cases-and particularly those with behavioural presentations-are familial, with up to c. The histopathological hallmarks are widespread cortical and subcortical gliosis, loss of large cortical nerve cells, and microvacuolation. The peak incidence of onset is 4565 years of age, but 10% or more may have onset after the age of 70. The gross pathological appearance in typical cases is of selectively atrophied frontotemporal regions which may be so severe as to produce the so-called knife-edged gyri, and deep widened sulci. Primary progressive aphasia in turn can be subdivided into progressive nonfluent aphasia, semantic dementia, and logopenic variants. There is often impaired judgement, an indifference to domestic and professional responsibilities, and a lack of initiative and apathy. Social skills deteriorate and there can be socially inappropriate behaviour, fatuousness, jocularity, and abnormal sexual behaviour with disinhibition. Many patients are restless with an obsessive-compulsive and ritualized pattern of behaviour, such as pacing or hoarding. Emotional lability, mood swings, and loss of empathy are common; psychiatric phenomena such as delusions and hallucinations are less common, although may be seen in some of the genetic forms, and in C9orf72 cases in particular. Patients often become rigid and stereotyped in their daily routines and food choices. Patients may show changes in music preference and altered sensitivity to pain and temperature. More detailed neuropsychological tests of frontal function (such as the Wisconsin Card Sorting Test or the Stroop Test) usually show abnormalities. Memory is relatively spared in the early stages, although it deteriorates as the disease advances. Primitive reflexes such as grasping and utilization behaviour develop during the disease process. Primary progressive aphasia-progressive nonfluent aphasia, semantic dementia, and logopenic aphasia In progressive nonfluent aphasia there is a gradual loss of expressive language abilities with impairments in motor speech impairment (apraxia of speech) and/or grammatical aspects of language production. This leads to nonfluent, agrammatical, and poorly articulated speech with phonological errors. Repetition of multisyllabic words and phrases is impaired but, at least early in the disease, word comprehension and object recognition are well preserved. In semantic dementia there is a profound loss of conceptual knowledge (or semantic memory), causing anomia and impaired comprehension of words, objects, or faces. Patients are unable to understand less frequent words and fail on a range of semantically based tasks such as matching words to pictures and matching pictures according to their meaning. Repetition of words and phrases is normal even though patients may be unaware of their meaning. Semantic knowledge is variably preserved, and while being able to pronounce individual words patients have grave difficulties repeating longer phrases or sentences, and show impaired working memory. The differences between the various syndromes described earlier may be clear early in the disease, but there is increasing overlap between the temporal and frontal syndromes as the disease progresses. Specific atrophy patterns in the correct clinical context may provide clues to a genetic cause: progranulin mutations are associated with often profound atrophy of one hemisphere, whereas mutations in tau are often associated with symmetrical inferior medial temporal atrophy. Management and prognosis There is no curative treatment at present, so the general management of the person with dementia and their family, as discussed earlier, is of prime importance. Shows progressive deterioration of behaviour and/or cognition by observation or history (as provided by a knowledgeable informant). Ascertainment requires that symptoms be persistent or recurrent, rather than single or rare events A. Early perseverative, stereotyped or compulsive/ritualistic behaviour [one of the following symptoms (D. Neuropsychological profile: executive/generation deficits with relative sparing of memory and visuospatial functions [all of the following symptoms (F. Exhibits significant functional decline (by caregiver report or as evidenced by Clinical Dementia Rating Scale or Functional Activities Questionnaire scores) C. Pattern of deficits is better accounted for by other nondegenerative nervous system or medical disorders B. Pattern of deficits is better accounted for by other nondegenerative nervous system or medical disorders 24. Prominent initial episodic memory, visual memory, and visuoperceptual impairments 4. Effortful, halting speech with inconsistent speech sound errors and distortions (apraxia of speech) At least 2 of 3 of the following other features must be present: 1. Imaging- supported nonfluent/agrammatic variant diagnosis Both of the following criteria must be present: 1. Impaired single-word comprehension At least 3 of the following other diagnostic features must be present: 1. Impaired object knowledge, particularly for low-frequency or low-familiarity items 2. Impaired repetition of sentences and phrases At least 3 of the following other features must be present: 1. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Patients with a family history of dementia should be considered for screening for tau, progranulin, or C9orf72 mutations after appropriate genetic counselling. The prognosis can be variable with different rates of progression between individuals. First, there is a tendency to marked spontaneous fluctuations in cognitive abilities, particularly alertness and attention. These fluctuations occur in 5075% of patients, may be profound, and may last anywhere between hours and several days. Second, visual hallucinations, illusions, and fleeting misidentification phenomena occur in at least two-thirds of patients even at an early stage and without drug provocation. The hallucinations are typically well-formed, silent, and are usually images of people or animals. In contrast to those seen in major psychiatric disease, these hallucinations are typically not threatening, and indeed may be reported as being comforting. In some cases, misidentification may lead to the Capgras phenomenon (the belief that someone close to them has been replaced by an exact double). Third is the occurrence of parkinsonism seen in 70100% of cases, which is usually mild in the early stages. In the later stages the akinetic rigid syndrome can cause severe disabilities in mobility and swallowing, and an increase in the number of falls. There is often an exquisite sensitivity to neuroleptic medication, which can in extreme cases precipitate the malignant neuroleptic syndrome (delirium, hyperpyrexia, muscle rigidity, massive elevation of creatine phosphokinase, and renal failure). Hallucinations may be spontaneous but may be associated with, or exacerbated by, dopaminergic medication. At least anecdotally, there are often marked improvements in hallucinations following treatment. There is some weaker evidence that memantine may provide benefits in these conditions. Vascular cognitive impairment Definition and epidemiology Recent years have seen significant changes in how we consider the impact that cerebrovascular disease may have on cognition. However, as the cognitive deficits produced by cerebrovascular disease may be both mild. In particular, disease of small perforating vessels (including not only lacunes in the basal ganglia and deep white matter, but also more diffuse lesions in the white matter, often termed leukoaraiosis) has emerged as a key mechanism of vascular cognitive impairment. Traditionally regarded as the second most common cause of dementia, it is difficult to estimate the true contribution of vascular disease to cognitive decline. Dopamine transporter imaging shows symmetrical reduced basal ganglia uptake (dot-like, rather than comma-like, in appearance) in a patient with clinically probable dementia with Lewy bodies. While there are some very rare genetic causes of Lewy body pathology which can have cognitive impairment (duplications/triplications of the -synuclein gene, mutations in the glucocerebrosidase gene), there is not a role for routine genetic testing. Management the treatment of patients with Lewy body disorders requires consideration of nonpharmacological approaches; minimizing the use of medications that may worsen the condition; and striking a balance between treating motor symptoms that may worsen cognition, and treatment for cognitive symptoms that may impact on mobility. As well as general nonpharmacological management of dementia, attention to any coexistent vision and hearing problems is important; ensuring appropriate lighting, especially at night, may improve hallucinations. It is important to review the need for, and where appropriate minimize or discontinue use of, drugs with anticholinergic actions which may worsen cognitive symptoms and postural hypotension. Antipsychotic drugs should be avoided wherever possible; if absolutely necessary, atypical antipsychotics should be used at the lowest possible dose and for the shortest duration possible. However, although dramatic motor improvements are not to be expected, a cautious medication trial is worth attempting if motor symptoms interfere with function. There may be severe language impairment, visuospatial disturbance, amnesia, and dyspraxia related to lesions in the middle and posterior cerebral artery distributions. A patient must have either developed dementia before, or within one year of the onset of parkinsonian syndromes. Cognitive deficits on either formal neuropsychological testing or a scale of global cognitive abilitiesa 4. Cognitive deficits are not sufficient to interfere significantly with functional independence, although subtle difficulties on complex functional tasks may be present Exclusion criteria 1. These criteria cannot be used for subjects who have an active diagnosis of drug or alcohol abuse/dependence. The diagnosis of dementia must be based on cognitive testing, and a minimum of 4 cognitive domains should be assessed: executive/attention, memory, language, and visuospatial functions. The deficits in activities of daily living are independent of the motor/sensory sequelae of the vascular event.
The addition of topical benzoyl peroxide can be used to try and eliminate resistant organisms diabetes diet quotes 1 mg repaglinide fast delivery. Hormonal therapies can help females with acne diabetes test how often purchase repaglinide no prescription, whether or not their serum androgen levels are normal diabetes vs hypoglycemia repaglinide 2 mg order with amex. Possible options are oestrogens diabetes insipidus subarachnoid hemorrhage cheap repaglinide 0.5 mg buy line, androgen receptor blockers diabetes type 1 research paper repaglinide 0.5 mg order amex, or agents designed to decrease the endogenous production of androgens by the ovary or adrenal gland. The oestrogen component of oral contraceptives increases sex-hormone binding globulin, thus decreasing free testosterone in healthy women. Oestrogens also decrease production of ovarian androgens by suppressing secretion of pituitary gonadotropins. The progestin component of oral contraceptives minimizes the risk of endometrial cancer. However, progestins like norethisterone have intrinsic androgenic activity so might aggravate acne. Drospirenone 3 mg combined with ethinylestradiol 30 µg has been shown to have a superior effect to a third generation-combined pill. The relative thromboembolic risk with cocyprindiol is slightly higher than that linked to non-antiandrogenic combined oral contraceptives but no higher than those containing third generation progestins. Side effects are dose-related and include potential hyperkalaemia, irregular menstrual periods, breast tenderness, headache, and fatigue. Although tumours have been reported in rodent models treated with spironolactone, this drug has not been directly linked with cancer in humans. There is a risk of feminization of a male fetus and thus pregnancy should be avoided. Isotretinoin is a synthetic form of vitamin A and is effective in severe inflammatory acne that has failed to respond to other treatments. Oral isotretinoin is the only agent that impacts on the four main aetiological factors driving acne. It is a lipid soluble drug, hence its absorption is enhanced when administered with food. Oral isotretinoin should not be combined with tetracyclines as both can lead to benign intracranial hypertension. Mucocutaneous problems are the most common side effect of oral isotretinoin, including cheilitis, irritant dermatitis, and blepharoconjunctivitis. Oral isotretinoin is a potent teratogen and women of childbearing age should not start therapy until a negative pregnancy test has been obtained, ideally two to three days prior to menstruation. Adequate contraception is essential for fertile, sexually active females before, during, and up to five weeks post-therapy. A recent European directive recommends mandatory pregnancy testing prior to the start of treatment and five weeks post-therapy and advocates monthly pregnancy testing throughout the treatment period. Baseline blood tests, including fasting lipids and liver function, should be done before starting therapy and are recommended at one month, then three-monthly throughout the treatment course. Adverse psychiatric events such as mood swings, depression, and suicidal ideation have been reported as possible idiosyncratic reactions to isotretinoin and must be highlighted. Epidemiological studies have not demonstrated a definite causal relationship between psychological effects and isotretinoin, but the association of depression with isotretinoin has not been satisfactorily investigated. Several small studies have trialled lasers, photodynamic therapy, and phototherapy with either clear blue or mixed blue-red light/radiation in inflammatory acne. Prognosis/outcome Acne is a chronic inflammatory skin disease which can result in physical and emotional scarring. Treatment regimens should be adopted to address as many aetiological factors as possible to optimize treatment results. Steroid acne has a monomorphic appearance and consists of noninflammatory and inflammatory lesions. Cosmetic acne Various cosmetic ingredients induce comedones, in particular lanolins, petrolatum, and certain vegetable oils. Patients develop inflammatory lesions, particularly on the cheeks, usually after three months of age. Treatment is similar to adult acne, but tetracyclines should be avoided due to the risk of discoloured teeth. Topical therapies and/or oral erythromycin (125 mg twice daily) or trimethoprim (100 mg twice daily) can be used safely. Gram-negative folliculitis this occurs as a complication of any long-term topical or oral antibiotic therapy. It is characterized by sudden onset of multiple pustules, often localized periorally and perinasally. This results from overgrowth of Gram-negative organisms including Escherichia coli, proteus, pseudomonas, and klebsiella. The offending antibiotic should be stopped and changed to oral trimethoprim or ampicillin. Acne conglobata this is an uncommon severe form of acne characterized by acne nodules, interconnecting sinuses, grouped comedones, and extensive scarring. Concomitant short courses of antibiotics and oral steroids might be required to control acute exacerbations. Associated systemic symptoms including fever, weight loss, arthralgia, and myalgia are evident. The aetiology is uncertain, but the presence of microscopic haematuria, erythema nodosum, increased response to P. Oral prednisolone is the treatment of choice followed by the cautious introduction of systemic isotretinoin. Several cases of acne fulminans have been triggered by anabolic steroids and testosterone. Pyoderma faciale this disorder is more common in adult women and often occurs in the context of emotional stress. These patients are not systemically unwell but the appearance of the disorder often adds considerably Special circumstances: Unusual acne variants Acne excoriée this occurs frequently in adolescent girls and young women. Treatment can be difficult, psychological problems should be investigated, and underlying acne lesions managed with standard acne treatment. The acne should be treated in the standard fashion and psychiatric collaboration is important. Treatment with prednisolone reducing over four to six weeks and the daily application of moderate to potent topical steroid for one week will help. Isotretinoin should be introduced after one week, and, if tolerated, can be gradually increased. It has been suggested that the depressed acne patient should be assessed for suicide risk. Understanding of pathophysiology allows topical and systemic therapies to be combined logically to target the individual aetiological factors. The possibilities for using immunomodulatory therapies and specific anti-inflammatory treatments are open to further developmental research studies and controlled trials. Other possibilities include insulin sensitizing agents, 5-reductase type 1 inhibitors, and possibly new anti-inflammatory agents such as lipoxygenase inhibitors. Post-inflammatory erythema or pigment changes may also result in visible abnormalities that are cosmetically unacceptable to patients. Acne scarring is a common consequence of acne and can occur, albeit mildly, in up to 90% of patients. A delay in appropriate acne management is more likely to result in significant scarring. Scarring commonly follows deep-seated inflammatory lesions, but can also occur as a result of more superficial inflamed lesions in scar-prone patients. Scars might show increased collagen (hypertrophic scars and keloids) or be associated with loss of collagen. Psychosocial effects of acne Studies have shown that many acne patients experience shame (70%), embarrassment and anxiety (63%), lack of confidence (67%), impaired social contact (57%) and significant problems with unemployment. When compared to other serious organic diseases, acne patients describe levels of social, psychological, and emotional problems as great as those reported with chronic disabling diseases such as asthma, epilepsy, back pain, arthritis, and diabetes. Clinical depression has been demonstrated in acne patients and the prevalence of active suicidal ideation is higher in acne patients than reported in the general population. The psychological impairment does not necessarily correlate with the clinical severity of disease. Widespread flat purpura without erythema should prompt a search for underlying haematological abnormalities such as platelet disorders. Larger (>1 cm) areas of purpura or bruising usually result from coagulation dysfunction. Palpable purpuric lesions, or those with a blanching component, suggest an associated inflammation as can be seen with vasculitis. A pressure ulcer (decubitus ulcer, bedsore, pressure sore) is due to localized injury to the skin and/or underlying tissue as a result of pressure alone, or in combination with shear and/or friction. The presence of moisture, particularly relevant in an incontinent patient, leads to a macerated (and therefore more vulnerable) skin. Consequences of post-thrombotic vein damage include further deep venous thrombosis, superficial thrombophlebitis, oedema, skin changes, and eventually ulceration. Approximately 70% of leg ulcers are venous in origin; the other 30% resulting from coexistent arterial disease, diabetes, and other skin disease. Most ulcers occur in the gaiter region at or above the level of the malleoli, where the persistently elevated ambulatory venous pressure has an adverse effect on the upstream capillary microcirculation. Nearly half of all venous ulcers are associated with deep vein valvular incompetence, usually secondary to previous deep venous thrombosis, while the remainder result from incompetence of the superficial or communicating veins (primary varicose veins). Introduction Vasculogenesis represents the formation of new blood and lymphatic vessels from endothelial precursors which share an origin with haemopoietic precursors. Adult bone marrow-derived haemopoietic cells extravasate around nascent vessels and stimulate growth of resident vessels by releasing angiogenic factors. These cells can also function as haemangioblasts, producing both haemopoietic and endothelial progenitors that give rise to new blood vessels (probably not lymphatic vessels). Angiogenesis is the growth of blood vessels through a process of sprouting and remodelling from existing vessels. The lymphatic system develops differently as most lymphatics differentiate from veins. Both blood and lymphatic vessels are crucial for organ growth in the embryo, as witnessed by mutations in some of the key genes in programming for cardiovascular and lymphatic development. The formation of blood and lymphatic vessels is a complex process controlled by numerous genes and molecular players. Close links exist between vessels and nerves; for example, axon-guidance signals such as ephrins and semaphorins allow vessels to navigate to their targets. Skin has been one of the most investigated tissues for understanding mechanisms of (lymph) angiogenesis, largely because it is so accessible. In some circumstances-for example, malignancy-the (lymph) angiogenesis activation becomes excessive and harmful so promoting the tumour growth and facilitating metastatic spread. Conversely, in arterial ischaemia the angiogenic switch is insufficient, preventing revascularization and healing of skin ulcers. In recent years, angiogenesis promoters and inhibitors have served as therapeutic targets. The blood supply has a generous reserve to meet the requirements of wounding and repair as well as thermoregulation. Skin disorders invariably involve the vasculature, if only because inflammation drives an increase in blood (and lymph) flow. Surface pressure, by emptying the compressible venules and veins and reducing capillary inflow, will blanch the skin. Purpura represents extravasation of red cells from microvessels into the dermis and cannot be blanched. Simple purpura is not raised, but if it is associated with inflammatory changes of the blood vessels (vasculitis), the mass of cells and oedema makes the purpura palpable. More extensive release of red cells into the skin and subcutis (haemorrhage) will result in a bruise (ecchymosis). Differences between purpura and bruising are simply a matter of degree or depth of haemorrhage. The cause may be due to: thrombophilia; excessive intravascular pressure; weakness of the blood vessel wall or surrounding stroma (as seen with steroid therapy). Gravitational forces, by increasing venous and consequently capillary pressure, are likely to make purpura more evident in the lower limbs. They appear on the skin and mucous membranes as small, dull red, linear, stellate, or punctate markings. Telangiectases (telangiectasias) represent expansion of pre-existing vessels without any obvious new vessel growth (angiogenesis). Unfortunately, clinical appearance may vary greatly according to the site, depth, and type of blood vessel involved. For example, the macular (flat) telangiectases seen in scleroderma, generalized essential telangiectasia, and portwine stain are produced by dilatation of the postcapillary venules of the uppermost vascular plexus in the dermis. The common raised cherry angioma (Campbell de Morgan spot) is produced by spherical and tubular dilatations of capillary loops in the dermal papillae. A spider angioma (spider naevus) represents high flow filling of surface capillaries by a single feeding dilated arteriole which, if blanched, will obliterate the whole spider naevus. The cutaneous lesions of hereditary haemorrhagic telangiectasia represent small arteriovenous anastomoses. Vasoconstriction to the skin results in desaturation of the slow flowing blood, leading to bluish (cyanotic) discoloration overlying the polygonal plexus of superficial venules and veins; warming restores normal flow and colour. If a similar reduction in flow occurs for pathological reasons, for example, intravascular thrombosis in antiphospholipid syndrome or vasculitis in polyarteritis nodosa, the mottling is fixed and broken up in pattern (livedo reticularis). Necrosis of the skin occurs following vascular occlusion due to intravascular coagulation, vasculitis, emboli, hyperviscosity syndromes, or vessel wall thickening. Purpura Simple macular purpura/petechiae Bleeding into the skin may occur for local reasons or as part of a systemic disorder (Box 23.
Discount 0.5 mg repaglinide otc. Bacteria which causes Urinary infection are inside our body | Doctor Naanga Eppadi Irukkanum | News7.
References
- Foerster O. Motorische felder und bahnen. In Bumke O, Foerster O (eds), Handbuch der Neurologie. Berlin: Verlag von Julius Springer, pp. 1-45, 1936.
- Chobanian AV, Bakris GL, Black HR, Cushman WC, Green LA, Izzo JL Jr, Jones DW, Materson BJ, Oparil S, Wright JT Jr, Roccella EJ; National Heart, Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee: The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure: The JNC 7 Report. JAMA 2003;289(19):2560-2571.
- Paul M, Bougouin W, Geri G, et al. Delayed awakening after cardiac arrest: prevalence and risk factors in the Parisian registry. Intensive Care Med. 2016;42:1128-1136.
- Christante D, Pommier S, Givi B, et al. Hepatic artery chemoinfusion with chemoembolization for neuroendocrine cancer with progressive hepatic metastases despite octreotide therapy. Surgery 2008;144(6):885-893.
- Shi Y, Xu X, Zhao Y, et al. Short-term surgical outcomes of a randomized controlled trial comparing laparoscopic versus open gastrectomy with D2 lymph node dissection for advanced gastric cancer. Surg Endosc 2018;32(5):2427-2433.
- Giles MF, Rothwell PM: Risk of stroke early after transient ischaemic attack: a systematic review and meta-analysis, Lancet Neurol 6:1063-1072, 2007.
- Kasinath BS, Corwin HL, Bidani AK, et al: Eosinophilia in the diagnosis of atheroembolic renal disease, Am J Nephrol 7:173-177, 1987.
- Plaut D. Platelet function tests. ADVANCE for the Administrators of the Laboratory, July 2003.