

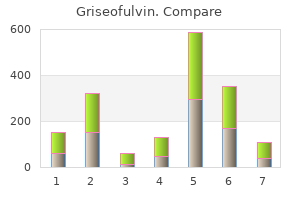
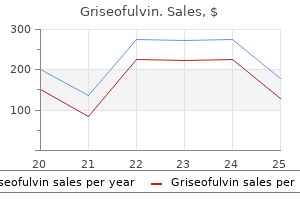
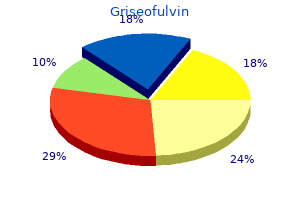
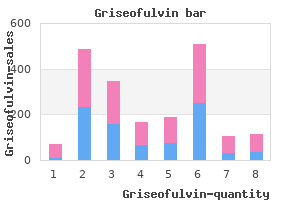
Griseofulvin
| Contato
Página Inicial
Joanna Chikwe, MD
- Assistant Professor
- Department of Cardiothoracic Surgery
- Mount Sinai Medical Center
- New York, New York
Metabolic effects of increasing doses of nitisinone in the treatment of alkaptonuria antifungal dog spray purchase genuine griseofulvin on-line. Ochronosis and alkaptonuria; report of a new case with calcified aortic valve stenosis diabet x antifungal skin treatment griseofulvin 250mg fast delivery. Routine neonatal screening programs have been most effective in the developed countries of the world ergot fungus definition buy griseofulvin cheap online. For these reasons antifungal treatments buy 250 mg griseofulvin with amex, the full-blown picture of the classic disease is rarely observed today in these countries antifungal foot spray 250 mg griseofulvin purchase with mastercard. The term "phenylketonuria" was first proposed by Penrose [2] who recognized the disease as the first in which there was a chemical cause of mental impairment. The brown eyes remind us that all patients with this disease do not have blue eyes. Routine neonatal screening had not yet been initiated in that country at the time of diagnosis. However, there is no amount of pigment in skin, hair, or irides that excludes the diagnosis. He had hypertonia and had a rapid unusual limping gait, in which he leaned forward to the left, toe-walked, swinging his right arm, and keeping the left at his side. Neurologic manifestations are not usually prominent, but about one third of the patients may have all of the signs of cerebral palsy [11]. Another one third of the patients have very mild neurologic signs, such as a unilateral Babinski response or hyperactive deep tendon reflexes. Another one third of untreated patients have no neurologic signs, except for mental impairment. Purposeless movements, rhythmic rocking, stereotypy, tremors, and athetosis may be seen. Hundreds of different alleles have been discovered, over 90 percent of which cause disease [16, 17], but only five are responsible for most human disease; the rest are rare. Almost two-thirds are missense mutations; 13 percent are small deletions; 12 percent splice-site mutations; six percent nonsense mutations; and one percent small insertions. Restriction enzyme polymorphism permitted heterozygote detection and prenatal diagnosis in the approximately 75 percent of families in which relevant polymorphism was identified [18]. Once the mutation in the phenylalanine hydroxylase gene is known, mutational analysis may be used for prenatal diagnosis and for heterozygote detection. In the best studied Northern European population, eight mutations have resulted in 64 percent of the mutant phenylalanine hydroxylase chromosomes [4, 5]. A number of the abnormal alleles identified have involved cytosine-phosphate-guanine (CpG) dinucleotides, which are known to be highly mutable. Expression of the mutant genes and assessment of enzyme activity in vitro has permitted correlations of phenotype with genotype [4]. Correlations with pretreatment concentrations of phenylalanine, and phenylalanine tolerance and the response to oral loading with protein were quite strong. Arginine to glutamine mutations in exons 5 and 7 were associated with variant phenotypes. Most of these have been missense mutations, such as the one that leads to complete inactivation of the enzyme in North African Jews [20]. A few deletions have been observed, such as the 22-bp deletion in exon 6 in an Arab family [21]. This has been demonstrated by liver biopsy [22] in which activity correlated well with in vitro expression analysis of the mutant gene. Correlations were also excellent in eight patients tested in vivo with deuterated phenylalanine [23]. Analysis of genotypephenotype correlations in an assembly of 365 patients reported [24], revealed a predominantly predictable or consistent phenotypic degree of severity in the majority. The severe mutation coded for a protein that was degraded faster than the milder variant. Heterozygosity has been demonstrated by assay of the enzyme in the liver, and of course, by mutational analysis. The reduction of this compound to reform tetrahydrobiopterin is catalyzed by dihydropteridine reductase [33, 34]. The synthesis of biopterin begins with guanosine triphosphate and proceeds through reduced neopterin (-D-erythro-7, 8-dihydroneopterin triphosphate) to a dihydro-precursor of tetrahydrobiopterin [35, 36]. The three isozymes have identical molecular weights and kinetic constants, but differ in charge [37, 38]. In the presence of a defect in phenylalanine hydroxylase, the first compound that accumulates is phenylalanine itself. There is a roughly linear relationship between the concentrations of phenylalanine in the blood and the urinary excretion of phenylpyruvic acid [44]. This is the compound that is responsible for the positive ferric chloride (FeCl3) test. Phenylpyruvic acid is subsequently converted to phenyllactic acid, phenylacetic acid, and phenylacetylglutamine. Phenylpyruvate is also hydroxylated in the ortho position, ultimately yielding orthohydroxyphenylacetic acid. There are a variety of secondary effects of the accumulation of phenylalanine and its metabolites. Decreased pigmentation has been related to the inhibition of tyrosinase by phenylalanine. Decreased levels of 5-hydroxytryptamine (serotonin) appear to be due to inhibition of 5-hydroxytryptophan decarboxylase by phenylpyruvic, phenyllactic, and phenylacetic acids. Decreased amounts of epinephrine, norepinephrine, and dopamine are presumably caused by inhibition of dopamine decarboxylase. This is accomplished by the routine screening of all infants for an elevated concentration of phenylalanine in the blood. It is generally carried out on discharge from hospital after the initiation of protein-containing feedings. A drop of blood collected from the heel on filter paper is analyzed for phenylalanine by the bacterial inhibition method developed by Guthrie and Suzi [46], or by a quantitative determination of the concentration of phenylalanine. A second positive is followed up with quantitative assay of the concentrations of phenylalanine and tyrosine in the blood confirming the phenylalaninemia and excluding transient tyrosinemia of the newborn, a common cause of a positive screening test. In the presence of an elevated concentration of phenylalanine and normal or reduced tyrosine, the patient may be admitted to hospital, where protein and phenylalanine intake are carefully monitored and fresh urine specimens collected. Precise determination of the concentration of phenylalanine in blood is of major importance. In this regard, the use of the Guthrie test is inappropriate, particularly in regions of the world where neonatal hepatitis and immature births are Diagnosis 121 table 15. When newborn screen positive: Obtain plasma for quantitative amino acids Begin low phenylalanine diet elevated Repeat Normal level <150 µmol/L no further control Plasma phenylalanine over 340 µmol/L (6 mg/dL). Delete Phenylalanine for: Monitor Plasma Quantative Phenylalanine* qd qd q13d q13d 4. Plasma Phenylalanine (mg/dL) Hours (µmol/L) 24 (410) 240605 48 (1020) 6051210 72 (2040) 12102420 96 (>40) >2420 *To prevent phenylalanine deficiency When plasma phenylalanine reaches the treatment range phenylalanine is added to the diet Individual amino acid requirements vary. The following are guidelines for initial dietary phenylalanine content dependent on the maximum pretreatment plasma levels: Plasma Phenylalanine (µmol/L) (mg/dL) Dietary Phenylalanine mg/kg 7. Monitor thereafter every week until 6 months old; q 2 weeks until 1 year old q 4 weeks until 3 years old q 6 months until 12 years old Yearly thereafter. A protocol for the management of the newborn detected by a positive screening test is given in Table 15. A conventional challenge in a three- to six-month-old infant is a 3-day intake of 24 oz of evaporated milk:water (1:1) which provides 180 mg/kg of phenylalanine. The challenge can be adjusted to 180 mg/kg of phenylalanine for an older, larger child. It is important to remember that this challenge was developed for use with infants, and the predominant experience is at the three-month level. In fact, symptomatic hypoglycemia and hyperinsulinemia have been reported in a 15-year-old patient so challenged [49]. Infants in whom this test did not yield levels higher than 1200 µmol/L [42] were classified as variants. Some variants represent molecular heterogeneity at the phenylalanine hydroxylase locus specifying variant enzymes with partial activity. Most of the variants have phenylalanine concentrations under 1200 µmol/L, and such infants can tolerate more than 75 mg/kg of phenylalanine per day. A small number of variant patients have been studied by liver biopsy [39, 42, 50], and in each a substantial defect in phenylalanine hydroxylase activity was demonstrated. Transient phenylalaninemia may represent an isolated delay in the maturation of phenylalanine metabolizing enzymes. It is because of this phenomenon that patients with phenylalaninemia are routinely tested for their dietary tolerance to phenylalanine during the first year of life. We strive to keep levels below 300 µmol/L and find that most patients in steady state do not approach any lower limit areas. The prevention of clinical disease by the restriction of dietary phenylalanine has provided the strongest evidence for the concept that the clinical manifestations of the disease result from the abnormal chemical milieu that follows the genetic defect. At a daily dose of 20 mg/ kg, kuvan can result in increased dietary tolerance for phenylalanine or, in rare instances, replacement of the phenylalanine-restricted diet. In a meta-analysis of published data, patients treated with sapropterin prior to six years of age were found to have cognitive performance no different from controls, despite ingestion of substantially more phenylalanine containing foods [55]. This requires the provision of enough phenylalanine to meet the normal requirements of this essential amino acid for growth. It also requires the frequent quantitative assessment of the concentration of phenylalanine in the blood (see Table 15. However, Smith and colleagues [51] have recommended a smaller window of between 120 and 300 µmol/L. There were significant differences in school performance as measured by the Wide Range Achievement Test. Mannerisms, hyperactivity, and signs of anxiety have been reported in eight-year-old children treated since early neonatal diagnosis, and those whose diet was less strictly controlled were twice as likely to display abnormal behaviors than those more strictly controlled [59]. Levels of phenylalanine were positively correlated to age and inversely related to dietary adherence. The exact biochemical mechanism underlying the subtle neurocognitive defects remains unknown. The best management to prevent later appearance of subtle neurologic defects remains early detection and vigorous treatment. If high levels of phenylalanine continue to impair myelinization, one might even expect treatment to be useful well up to puberty, since myelination at least in the formatio reticularis is not finished at eight years of life. In addition, the effect of high concentrations of phenylalanine on synaptogenesis is not known. Some older patients find that their skin feels better with modest restriction of phenylalanine. In any case, the rigidity with which one controls the level of phenylalanine can probably be relaxed after six years of age, but it is prudent to continue some restriction in the intake of phenylalanine. Patients were advised to continue dietary restriction for life, but only ten followed this regimen. Intellectual outcome appears to have best been predicted by the presence or absence of early insult to brain, while performance on a test of problem-solving correlated best with concurrent levels of phenylalanine even in adulthood. In a recent study of 95 patients treated from the neonatal period and assessed at 12 years of age, best cognition results were those of patients whose phenylalanine values were kept consistently below 900 µM. This was the case in those less than two years of age managed with a restricted diet. The level of prealbumin has been found [68] to correlate well with protein adequacy in this disease, and the threshold level is 20 mg/dL. Linear growth can be expected to be impaired in patients with levels lower than 20 mg/dL. Decrease in glomerular filtration rate, proteinuria and hypertension were found in patients 1543 years of age [69]. The high lifelong intake of mixtures of amino acids was thought to have been nephrotoxic. These findings are dramatically reduced when maternal phenylalanine levels are well maintained between 120 and 360 µM. Other studies have also indicated that if the metabolic control is started before eight weeks of pregnancy, a better result is obtained. Abnormalities were significantly lower when the treatment of the mother was initiated before eight weeks of pregnancy [71]. A survey of male patients over 18 years in the United States identified 40 men who had 64 children, but did not yield data on fertility rate [73]. The variability in manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria). The phenylalanine hydroxylase locus: a marker for the history of phenylketonuria and human genetic diversity. Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria. A missense mutation S349P completely inactivates phenylalanine hydroxylase in North Africa Jews with phenylketonuria. Molecular genetics of phenylketonuria in Mediterranean countries: a mutation associated with partial phenylalanine hydroxylase deficiency. Sensitive in vivo assay of the phenylalanine hydroxylating system with a small intravenous dose of heptadeutero L-phenylalanine using high pressure liquid chromatography and capillary gas chromatography/mass fragmentography. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotypephenotype correlations. Biochemical characterization of mutant phenylalanine hydroxylase enzymes and correlation with clinical presentation in hyperphenylalaninaemic patients. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Sapropterin: a review of its use in the treatment of primary hyperphenylalaninaemia.
Classic Celiac Trunk: Hepato-GastroSplenic Trunk this is the classic configuration of the celiac trunk fungus gnats australia buy griseofulvin online pills, with the hepatic fungus gnats elimination discount griseofulvin 250 mg with mastercard, left gastric and splenic artery arising as a trunk from the abdominal aorta antifungal mouth discount griseofulvin online american express. Three subtypes are described: (1) hepatosplenic trunk with the left gastric artery arising from the trunk antifungal cream in ear 250mg griseofulvin purchase with amex. Hepatosplenic Trunk the hepatic and splenic arteries form a trunk antifungal underarm purchase griseofulvin online, and the left gastric artery arises from the aorta. Hepatogastric Trunk the hepatic and left gastric arteries arise from a common trunk, and the splenic artery arises directly from the aorta. Hepato-Splenic-Mesenteric Trunk the left gastric artery arises directly from the aorta and the hepatic, the splenic, and the mesenteric arteries form a single trunk. The anterior branch crosses anteriorly to the head of the pancreas joining the anterior pancreaticoduodenal arcade. The posterior branch crosses posteriorly to the head of the pancreas anastomosing with the posterior pancreaticoduodenal arcade. When the middle hepatic artery does not exist, it is replaced by the right or left or both at the same time. These branches vascularize the jejunum and ileum, except for the terminal ileum, forming a series of arches and anastomoses in three or four levels. The straight arteries (vasa recta) and the short arteries (vasa brevia) arise from the 4th- or 5th-level arches and supply the intestinal wall and mucosa. Celiac-Colic Trunk the left colic artery or the middle colic artery arises from the celiac trunk. Absent Celiac Trunk the three main arteries arise directly from the abdominal aorta. It is oriented to the right underneath the retroperitoneal layer and vascularizes the terminal ileum, the right colon, the cecum, and the appendix. This artery supplies all of the small intestine, the right colon, and most of the transverse colon. The descending anastomoses with the ileocolic artery and the ascending with the middle colic artery. The right anastomoses with the right colic artery, the left branch with the left colic artery, a branch of the inferior mesenteric artery through the marginal arteries. It may be replaced and arises from the dorsal pancreatic artery or from the celiac trunk. At the tip of the villus, the arteriole is arborescent and divides into a fine subepithelial channel system, which eventually drains into a central venule. The arterial and venous loops in the villi are so close that there is evidence of a countercurrent exchange of oxygen and nutrients in the intestinal villus, aggravated in low flow states as in intestinal ischemia. There is evidence of arteriovenous anastomoses in the submucous plexus of the gut wall, better demonstrated in the stomach and colon rather than the small bowel. The regulation of the blood flow reaching the intestine is complex and interdependent, playing an important role in the theory of vascular circuits running in parallel, with resistance in series. The replaced hepatic artery or accessory hepatic artery follows a path behind the head of the pancreas, in intimate contact with the pancreas tissue, sometimes actually encircled by pancreatic parenchyma. The arc of Bühler is an uncommon direct pathway between the celiac and superior mesenteric arteries, due to the remaining embryologic ventral anastomosis. The inferior mesenteric artery supplies the left third of the transverse colon, the descending colon, the sigmoid colon, and part of the rectum. It follows a retroperitoneal path in the left colonic branches and enters the sigmoid mesocolon with the rectal arteries. Left Colic Artery this artery courses a retroperitoneal route and divides into ascending and descending branches. The ascending branch reaches the transverse mesocolon where it anastomoses with the middle colic artery. Arches that originate from this ascending branch provide the blood supply of the distal transverse colon and descending left colon. From the anastomosis between these arteries, the continuous marginal artery near the colon wall originates; this artery is called the marginal artery of Drummond. The anastomosis of the marginal artery with the ascending branch of the left colic artery and the distal left middle colic artery is frequently made through an additional arcade, the arc of Riolan. Terminal Arteries and Intestinal Villi the intramural vessels of the bowel arise from both vasa recta and the vasa brevia and form the external muscular plexus. After crossing the muscle, a rich submucosal plexus is formed from which a few recurrent branches rim backward into the muscle and may join the external muscular plexus. The submucosal plexus is found over the length of the bowel and is arranged in a coarse rectangular pattern. From the submucosal plexus arterioles spring the vertical arterioles, thereby reaching the villi and the mucous membrane. The circulation to the villus is supplied from a single arteriole, about 20 mm in diameter, which originates from the submucosal plexus and runs up to the villi stroma and Chapter 18 Abdominal Aorta and Branches 467 Sigmoid Arteries There are two or three sigmoid arteries within the sigmoid mesocolon. There is an upper anastomosis with the descending branch of the left colic artery and a lower anastomosis with the superior rectal artery. Superior Rectal Artery this artery descends into the pelvis in the sigmoid mesocolon and when it reaches the rectum divides into two lateral branches, right and left, dividing further into smaller branches reaching the sphincter ani internus, forming loops around the lower rectum. There are communications with the middle rectal artery (branch of the internal iliac artery) and with the inferior rectal artery (branch from the internal pudendal artery). At the internal aspect of the zona reticularis, there are muscle fibers, restricting or releasing the flow, acting as a dam at the corticomedullary junction. Some larger arterioles bypass the described route and connect the capsular artery with the medullary capillaries. There is a medullary plexus that connects with the medullary vein, which emerges from the hilum to form the suprarenal vein, draining to the inferior vena cava in the right and to the renal vein in the left side. Renal Arteries Extrarenal Arteries Each renal artery is described as a single vessel, emerging on each side of the vertebral column between the first and the second lumbar vertebra, immediately below (1 to 2 cm) of the superior mesenteric artery. The renal artery usually has a slightly oblique cranialcaudal course, and close to the renal sinus, after giving off the inferior suprarenal artery, it divides into an anterior and a posterior branch. The aortic origin of the left renal artery is higher than the right renal artery origin; nevertheless, it is not a definitive rule. Multiplicity of renal arteries is more common than multiple veins and is more prevalent than any other arteries of the same size in other organs. Material of Investigation Renal pedicles (266) were analyzed that were dissected from 133 formalin-fixed cadavers of adult patients, of both sexes, who died of causes not related to the urinary tract. The current nomenclature used and that must be adopted to denominate the renal arteries is the following: Hilar Artery Aortic branch that penetrates the kidney in the hilar region. Extrahilar Artery Renal artery branch that has an extrahilar penetration (superior pole) in the kidney. Superior Polar Artery Aortic branch that penetrates the kidney in the superior pole. Inferior Polar Artery Aortic or common iliac artery branch that penetrates the kidney in the inferior pole. Precocious Bifurcation Renal artery in which the main trunk has less than 1 cm in length before branching off. When variations in the renal arteries exist, these vessels shall be named multiple arteries; the words "extra," "aberrant," and "accessory," should be avoided because these vessels are normal segmental end arteries, without anastomoses between them. Even the term "supernumerary" should not be used because it might imply that these vessels are superfluous. Lateral Branches of the Abdominal Aorta Inferior Phrenic Artery the inferior phrenic arteries may arise together as a trunk or separately as independent vessels, just above or at the origin of the celiac trunk. The medial curves forward and the lateral reaches the thoracic wall, having anastomosis with the posterior intercostal and musculophrenic arteries. Each artery gives the small superior suprarenal branches to the upper portion of the adrenal glands. The inferior phrenic artery supplies the Glisson capsule of the liver through anastomoses at the bare area of the liver within the triangular ligaments. Arteries of the Adrenal Glands the adrenal glands are vascular organs and are supplied by three groups of arteries. The capsular arteries ramify extensively before entering the gland to form a subcapsular plexus, which extends into the zona glomerulosa as sinusoids. The sinusoids continue into the zona fasciculata as straight cortical sinusoids between 468 Atlas of Vascular Anatomy the presence of the superior pole extrahilar branch. There was no significant statistical difference in arterial variations between right and left kidneys. In addition, in individual cases, the number of multiple arteries (when present) tends to be greater in black subjects than in white subjects. Angle of the Origins of the Renal Arteries In the transverse plane, the location of the origin of the right renal artery tends to be anterolateral, with an angle ranging from 0° to 70°, and the average angle is 30° to 35°. The left renal artery tends to be posterolateral or lateral, with an angle ranging from -50° to 35°, and the average angle is -11° to 15°. The afferent arteriole invaginates into the glomerular capsule (Bowman), forming the glomerulus, from which an efferent arteriole leaves. The complex formed by the glomerulus invaginated into the capsule is called renal corpuscle (Malpighi). In the renal corpuscle, one may identify a vascular pole (site where the afferent and efferent arterioles are located) and, in a diametrically opposite position, a urinary pole (site where the glomerular capsule narrows into a tube). Because the division branches of the renal artery and the intrarenal distribution of these vessels vary, study of the isolated vessels has a limited practical value. An analysis considering the anatomic relationship between the intrarenal arteries and the kidney collecting system, and considering these relationships in the specific kidney regions, is useful. Moreover, the anatomic relationships between the arteries and the collecting system have a fairly constant pattern in the different kidney regions. Also, these relationships are independent of both the number and the arrangement of the arterial segments. Material of Investigation Eighty-two three-dimensional endocasts of the renal collecting system together with the intrarenal arteries, obtained from 41 male and female fresh cadavers from patients who died of causes not related to the urinary tract, were studied. A yellow polyester resin was injected into the ureter to fill the kidney collecting system, and a red resin was injected into the main trunk of the renal artery to fill the renal arterial tree, according to the proportions and technique described previously. To study the intrarenal arteries and their anatomic relationships to the collecting system, the arterial tree was injected to its full capacity to preserve the same spatial associations as existed in vivo. Because the polyester resin has low viscosity and consequently high penetration power, the injection reached the interlobular arteries, the afferent arterioles, the glomerular tufts, the efferent arterioles, and even the vasa recta. Therefore, after organic matter corrosion and washing of the injected specimen, the cast looks like a sponge. If one examines the cast under magnification, it is easy to identify the microcirculation of the kidney. The fine vessels and glomerular tufts were removed by needle handpicking, allowing clear visualization of the targeted arteries and the underlying collecting system. Findings the findings were presented in relation to the pelvicaliceal system and considering the specific kidney regions. It is assumed that caudal ectopic kidneys have multiple arteries, but it is not always the case. The most common type of fused kidney is the horseshoe kidney, but a variety of ectopic kidneys exist, including crossed ectopia, fused presacral kidney, and pelvic kidney. Multiple arteries originating from the lower aorta or pelvic arteries are usually present to supply the ectopic kidney. Intrarenal Arteries the main renal artery divides into an anterior and a posterior branch after giving off the inferior suprarenal artery. Whereas the posterior branch (retropelvic artery) proceeds as the posterior segmental artery to supply the homonymous segment without further significant branching, the anterior branch of the renal artery provides three or four segmental arteries. The renal artery branches are terminal vessels, and there is no anastomosis between the segmental arteries. These segmental arteries divide before entering the renal parenchyma into interlobular arteries (infundibular arteries), which progress adjacent to the caliceal infundibula and the minor calices, entering the renal columns between the renal pyramids. As the interlobar arteries progress, they give origin (usually by dichotomous division) to the arcuate arteries. The arcuate arteries give off the interlobular arteries, which run to the periphery, thereby giving off the afferent arterioles of the glomeruli. Superior Pole the superior segmental artery (apical) can have different origins, but it usually arises from the anterosuperior segmental artery, being positioned in the medial midline and progressing Chapter 18 Abdominal Aorta and Branches 469 to the uppermost region of the superior pole. This artery often has a proximal origin, passes far from the upper infundibulum to reach the superior segment (apical). The upper caliceal group was involved by these two arterial trunks, which coursed alongside the anterior and posterior surfaces of the caliceal infundibulum. In these cases, the anterior aspect of the inferior pole is supplied by the anterior branch and its posterior aspect is supplied by the posterior branch. Dorsal Kidney the dorsal kidney is supplied by the posterior segmental artery (retropelvic artery), which is a direct extension of the posterior division of the main renal artery. Within the kidney substance, the retropelvic artery usually describes an arc and from its convexity three constant subdivision branches emerge and may be identified (superior, middle and inferior;. The superior branch is close to the posterior surface of the upper caliceal infundibulum. In some instances, the posterior segmental artery may give off two, or even three, superior branches related to the dorsal aspect of the superior pole. The middle branch supplies the middle portion of the posterior segment and may interdigitate with the anterior branches of the mid-kidney. Its course and arrangement is dependent on the inferior vascularization, as described previously. The posterior segmental artery itself was in close relationship to the upper infundibulum or to the junction of the pelvis with the upper calix in 57. In this situation, this artery usually described an arc that contacted the upper infundibulum. Midzone (Hilar) In all cases, the arterial supply to the anterior surface of the kidney midzone arose from the anterior division of the renal artery.
Definitive testing for activity can be accomplished in cultured fibroblasts anti fungal infection medicine order griseofulvin with a mastercard, lymphoblasts fungus evolution griseofulvin 250mg buy cheap, or freshly isolated lymphocytes fungus mushroom order cheap griseofulvin on-line. Large doses (up to 20 mg/kg) are required in the absence of this recycling enzyme [82] anti-fungal remedies for dogs order cheap griseofulvin on line. In addition to controlling the concentration of phenylalanine fungus wood treatment discount griseofulvin online, treatment of this condition must correct deficiencies of neurotransmitters. In some patients, a fixed preparation of carbidopa may have to be altered to give more or less carbidopa. The level of prolactin in serum is References 137 elevated in these diseases, which is an index of abnormal dopamine homeostasis, and it may be useful to monitor levels in order to guide therapy [83]. Folinic acid has to be employed in doses of 1520 mg/day in one to two doses [64, 81, 88]. Progressive improvement, with disappearance of myoclonus, involuntary movements, and tetraplegia, has been reported following treatment with biogenic amine precursors [4, 6]. There is evidence that early treatment may prevent progression [6, 24], but overall experience indicates that prognosis should be guarded, especially if not treated as newborns. This uncovered deficits in executive function, which has been thought to be controlled by prefrontal dopaminergic systems. All patients with the mild form were neurologically normal, except for one with transient dystonia, while all with the severe form had impressive neurologic impairment. A good result is by no means guaranteed even if treatment follows all of the guidelines. The first led to a girl who was said to have above average intellectual development despite a left hemiparesis and absence of the corpus callosum and right-sided schizencephaly. The second, a boy, had no anatomic abnormalities and was described as normal at five years of age. A new cofactor required for the enzymatic conversion of phenylalanine to tyrosine. Atypical phenylketonuria accompanied by a severe progressive neurological illness unresponsive to dietary treatment. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. Neonatal hyperphenylalaninemia presumably caused by a new variant of biopterin synthetase deficiency. Variant of dihydropteridine reductase deficiency without hyperphenylalaninemia: effect of oral phenylalanine loading. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterindependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Tetrahydrobiopterin deficiencies without hyperphenylalaninemia: diagnosis and genetics of dopa-responsive dystonia and sepiapterin reductase deficiency. Phenylalanine loading in pediatric patients with dopa-responsive dystonia: revised test protocol and pediatric cutoff values. Progression of 6-pyruvoyl-tetrahydropterin synthase deficiency from a peripheral into a central phenotype. Dihydropteridine reductase deficiency diagnosis by assays on peripheral blood cells. Guanosine triphosphate cyclohydrolase I deficiency: early diagnosis by routine urine pteridine screening. Antenatal diagnosis of tetrahydrobiopterin deficiency by quantification of pterins in amniotic fluid and enzyme activity in fetal and extrafetal tissue. Neonatal hyperphenylalaninemia presumably caused by guanosine triphosphate cyclohydrolase deficiency. Chromosomal location of two human genes encoding tetrahydrobiopterin-metabolizing enzymes: 6-pyruvoyl-tetrahydropterin synthase maps to 11q223-q233 and pterin-4a-carbinolamine dehydratase maps to 10q22. Mutation in the 4a-carbinolamine dehydratase gene leads to mild hyperphenylalaninemia with defective cofactor metabolism. New variant of phenylketonuria with progressive neurological illness unresponsive to phenylalanine restriction. Biopterin-dependent hyperphenylalaninemia due to deficiency of 6-pyruvoyl tetrahydropterin synthase. Dihydropteridine reductase deficiency associated with severe neurologic disease and mild hyperphenylalaninemia. Atypical (mild) forms of dihydropteridine reductase deficiency: neurochemical evaluation and mutation detection. Atypical phenylketonuria with dihydrobiopterin synthetase deficiency: absence of phosphate-eliminating enzyme activity demonstrated in liver. Peripheral tetrahydrobiopterin deficiency with hyperphenyl-alaninemia due to incomplete 6-pyruvoyl tetrahydropterin synthase deficiency or heterozygosy. Neonatal hyperphenylalaninemia presumably caused by guanosine triphosphatecyclohydrolase deficiency. Guanosine triphosphate cyclohydrolase I assay in human and rat liver using high-performance liquid chromatography of neopterin phosphates and guanine nucleotides. Identification of mutations causing 6-pyruvoyl-tetrahydropterin synthase deficiency in four Italian families. Single-step mutation scanning of the 6-pyruvoyl-tetrahydropterin synthase gene in patients with hyperphenylalaninemia. Tetrahydrobiopterin biosynthesis defects examined in cytokine-stimulated fibroblasts. A missense mutation in a patient with guanosine triphosphate cyclo-hydrolase deficiency missed in the newborn screening program. Tetrahydrobiopterin non-responsiveness in dihydropteridine reductase deficiency is associated with the presence of mutant protein. Molecular and immunological comparison of human dihydropteridine reductase in liver cultured fibroblasts and continuous lymphoid cells. Dihydropteridine reductase deficiency-D identification of natural mutations and analysis by recombinant expression and in vivo protein studies. Evaluation of a fetus at risk for dihydropteridine reductase deficiency by direct mutation analysis using denaturing gradient gel electrophoresis. Primapterin anapterin and 6-oxo-primapterin three new 7-substituted pterins identified in a patient with hyperphenyl-alaninemia. Dihydropteridine reductase deficiency: non-response to oral tetrahydrobiopterin load test. Diagnosis of tetrahydrobiopterin deficiency using filter paper blood spots: further development of the method and 5 years experience. Estimation of tetrahydrobiopterin and other pterins in cerebrospinal fluid using reversed-phase high-performance liquid chromatography with electrochemical and fluorescence detection. Unsolved problems in diagnosis and therapy of hyperphenylalaninemia caused by defects in tetrahydrobiopterin metabolism. Phenotypic variability, neurological outcome and genetics background of 6-pyruvoyltetrahydropterin synthase deficiency. Outcome and long-term follow-up of 36 patients with tetrahydrobiopterin deficiency. Maternal tetrahydrobiopterin deficiency: the course of two pregnancies and follow-up of two children in a mother with 6-pyruvoyltetrahydropterin synthase deficiency. Autoradiographic distribution of [14C]tetrahydrobiopterin and its developmental change in mice. There is extreme irritability, hypokinesia, severe progressive developmental delay, and progressive neurologic dysfunction. Extrapyramidal movement disorder, comprising parkinsonismdystonia, is obvious from early childhood. Physical examination may reveal brisk knee jerks and extensor plantar responses [6]. Before the definitive diagnosis is established, oculogyric crises are often mistaken for seizures, and multiple antiepileptic therapies are initiated, all with little effect on these dopamine deficiency phenomena. Benzodiazepins may ameliorate or stop oculogyric crises, as well as paroxysmal dystonia. He had severe truncal hypotonia and rigidly of limbs, as well as oculogyric crises. Providing enough energy and fluid is often a problem, and patients are at risk of becoming cachectic. Additional autonomic manifestations include excessive sweating and temperature instability. Sleeping disturbances and episodes of excessive crying can be very troublesome and cause demanding problems for families and caregivers. Reduced catecholamine production puts patients at risk for apnea and/or sudden cardiac arrest, especially when the patient is exposed to stressful situations including hospitalization for diagnostic procedures or surgery [8]. Especially in younger children, hypoglycemia can be a problem and in addition orthostatic hypotension when they become older. Insufficient production of growth hormone and response to it provide factors which contribute to hypoglycemia. Patients usually remain significantly below their target height, often below the 3rd percentile. Patients are socially interactive, though impaired mental or motor development, or both, may be major features of the disease (63 percent) [3]. In many cases, cognitive evaluation is not possible because of the severe motor impairment, and cognitive function is always much less impaired than motor functions. In one patient hypotonia, short periods of hypertonicity, and oculogyric crises were present but, even without treatment, the child could sit and communicate nonverbally by the age of two years [9]. Three patients (two siblings and a cousin) had syndromic features as well as intellectual disability. They showed craniofacial dysmorphisms, chronic diarrhea with or without recurrent hypoglycemia, gastroesophageal reflux and progressive kyphoscoliosis. They did not display 144 Biogenic amines Seizure types are grand mal or complex focal seizures. It is sometimes difficult to discriminate seizures from oculogyric crises and dystonic spasms. Psychiatric disorders in carriers In several series, there was an elevated incidence of psychiatric disorders in relatives of 1st or 2nd degree [3, 11, 12]. These psychiatric disorders were depression, psychosis, suicide or suicide attempts. It is necessary to follow a strict protocol for the collection of samples, with special tubes and prompt shipment to the referral laboratory overnight on dry ice [14]. Age-related reference values are critical, as well as recognition of the rostrocaudal pattern of concentrations. Craniofacial dysmorphism include elongated face with long nose, high arched palate, large open mouth with thick lips, prominent chin, malar hypoplasia. Measurement of the urinary concentrations of the neurotransmitters themselves might lead to (paradoxically) normal findings [16]. Finally, prolactin secretion by the pituitary is regulated by neurosecretory dopamine neurons in the hypothalamus. Dopamine inhibits secretion, so measurement of an increased serum prolactin may provide a clue to the diagnosis. The diagnosis can also be confirmed by assay of enzyme activity in biopsied liver [1, 2]. Preimplantation and prenatal genetic diagnosis has also been accomplished using an amplification refractory mutation system-quantitative polymerase chain reaction technique [17]. If the mutation can be detected, this will provide an ideal method for prenatal diagnosis and heterozygote detection. Dopamine is synthesized in the substantia nigra, ventral tegmentum, and hypothalamus. Its deficiency affects voluntary movements, cognitive function, and emotion, but also hormonal functions. Dopamine deficiency results in progressive extrapyramidal movement disorders, especially hypokinesia, parkinsonismdystonia and chorea. Dopamine does not only play a central role in the control of movement, but is also relevant for cognition, emotion/affect, and neuroendocrine, pituitary gland hormones (prolactin and growth hormone). Under physiologic circumstances, dopamine concentrations are also high in the kidney, where it is involved in control of sodium and water transport. Reduction or inappropriately low response of norepinephrine and epinephrine can lead to hypoglycemia, ptosis, and autonomic disturbances with loss of regulation of body temperature, vascular tone and blood flow, decreased blood pressure, and inappropriate response to stress. Deficiency of serotonin appears not to lead to severe neurologic symptoms, but affects appetite, sleep, memory, learning, mood, and modulation of pain mechanisms, as well as cardiovascular and endocrine functions. No systematic studies on drug therapy are available, and there are no clear guidelines or therapeutic recommendations. Striking improvement in tone and mobility was reported [1, 2] in the twins initially reported following treatment with a monoamine oxidase inhibitor, a dopamine antagonist, and pyridoxine. Medication with documented success in at least some patients are compiled in Table 17. All of them have to be prescribed, "off-label" and without established pediatric dosage regimens. A suggestion of sex differences was found [5] in a series of five male patients who responded well to treatment and progressed developmentally while five females and two males responded poorly. About half of the patients stabilize on individual treatment regimens and achieve different degrees of motor and psychosocial skills. About 20 percent improve to the point that they successfully manage regular schooling as long as some help is provided for the remaining motor handicap. Despite therapeutic interventions, the disease course is often severe and may be fatal at a young age [3, 21].
In general cinnamon for fungus gnats purchase 250mg griseofulvin visa, haloperidol and droperidol have a lower incidence of hypotension than do lower-potency neuroleptic agents anti-yeast or antifungal cream buy 250 mg griseofulvin with visa. It is categorized as a high-potency neuroleptic because of its strong antidopaminergic activity fungus killing foods proven 250mg griseofulvin. The antidopaminergic activity is responsible for both its intended effects against delusions fungus gnats cannabis symptoms best order griseofulvin, hallucinations fungi definition biology online cheap griseofulvin online visa, and psychomotor agitation and its unintended parkinsonian symptoms. Administration of haloperidol both alone and in combination with benzodiazepines has been evaluated in a large number of clinical trials. However, two large retrospective studies encompassing more than 15,000 patients failed to demonstrate increased morbidity or mortality associated with the use of droperidol for the management of acute agitation. They concluded that droperidol is a safe drug when used at the recommended dosage. Its onset of action is 3 to 10 minutes with a peak clinical effect achieved in 30 minutes. The elimination half-life is 2 to 4 hours, but the sedative effects of droperidol may last up to 12 hours. This combination makes them an excellent choice for tranquilization of agitated patients. Because of a desirable safety profile, benzodiazepines are an excellent choice for sedating medically undifferentiated patients. Benzodiazepines can be used as single agents or in combination with an antipsychotic drug. Because of the possibility of respiratory depression, use benzodiazepines with caution in patients in respiratory distress or identified pulmonary pathology like obstructive sleep apnea. Additionally, hypotension, deep sedation, and paradoxical agitation have been reported. However, when administered in the doses recommended for agitation, adverse events are rare, thus making benzodiazepines the drugs of choice in most circumstances. Respiratory compromise is dose dependent and typically occurs only in the presence of other respiratory depressants. In healthy patients, particularly those suffering from agitated delirium, respiratory depression is very unlikely to occur, even when large doses of benzodiazepines are used. One difference that does bear mentioning is the use of propylene glycol, a solvent needed to keep nonwater-soluble benzodiazepines. In large doses, propylene glycol can cause an increased osmolar gap and metabolic acidosis and may precipitate or contribute to hypotension in some patients. This does not require involvement of the cytochrome P-450 system, so lorazepam has few drug-drug interactions. The studies also noted that midazolam, as expected, has a shorter duration of sedation. Midazolam is hydroxylated by the cytochrome P-450 system to its primary metabolite, -hydroxymidazolam, which undergoes glucuronide conjugation before being excreted in urine. Its duration of action is between 30 and 120 minutes and does not vary significantly by route of administration. No maximum dose has been established, and doses of up to 2000 mg have been used safely over a 24-hour period in patients experiencing delirium tremons. Following the administration of diazepam, peak sedation is reached in approximately 5 to 6 minutes, which allows additional titrated doses if the desired clinical effect is not achieved initially. This is particularly true for patients with a history of mental illness, for whom this class of medications is now considered appropriate first-line management. In a double-blind, randomized study, Martel and associates noted that ziprasidone was as effective as midazolam and droperidol in controlling acute agitation. Patients receiving ziprasidone and droperidol took longer to be sedated (30 minutes as compared with 15 minutes for midazolam) but were more deeply sedated at 60 and 120 minutes. Olanzapine reaches peak plasma concentrations in 15 to 45 minutes and has an elimination half-life of 21 to 54 hours. The drug is metabolized via direct glucuronidation and cytochrome P-450mediated oxidation. For example, ketamine has been used successfully in aeromedical transport as a sedative agent for patients with agitation. The anterior aspect of the thigh is a preferred site of injection for rapid tranquilization. Choosing the Best Agent Undifferentiated Agitation Aripiprazole Aripiprazole is an atypical antipsychotic that acts at both serotonin and dopamine receptors. Peak serum levels occur within 1 to 3 hours, with an elimination half-life of approximately 75 hours. Dissociative Agents Ketamine is a dissociative agent that has been used safely throughout the world for major surgery and with minimal monitoring. It inhibits the reuptake of catecholamines promoting bronchodilation and increases in both heart rate and blood pressure. Commonly raised as a caution, there is no proven issue with ketamine causing harmful increased intracranial pressure. Chan and associates demonstrated the combination of an antipsychotic and midazolam had a shorter time to sedation than midazolam alone. Administering escalating doses of benzodiazepines is a prudent choice in such circumstances when the clinician is comfortable prescribing a drug from this class. For patients who are suspected of intoxication from alcohol or other sedative agents, haloperidol, droperidol, or ziprasidone will provide rapid, safe, and effective tranquilization. If rapid sedation is required, typical antipsychotics or benzodiazepines should be used as first-line therapy. If the patient is frail or elderly or is known to have renal impairment, consider using smaller doses of a single agent. Continued use (> 8 to 10 weeks) of atypical antipsychotic agents has been associated with increased rates of death in cases of dementia-related psychosis. There are also electrical weapons that cause intense pain without incapacitating the target, so-called drive stun devices. A complete discussion of this topic is beyond the scope of this chapter, but it has been well reviewed elsewhere. The electrode-tipped barbs are attached to the electric device via two thin 21-foot wires and are similar in size to a No. The barbs may attach to clothing and fail to penetrate the skin, or they may become embedded in skin and must be removed. Agitation Caused by an Underlying Psychiatric Disorder Patients with an established psychiatric history and agitation attributed to schizophrenia, schizoaffective disorder, or the manic phase of bipolar disorder may be treated with typical antipsychotic agents, atypical antipsychotic agents, or benzodiazepines. However, a growing body of evidence seems to support the use of atypical antipsychotic agents in this circumstance. Although droperidol is a highly effective drug for rapid sedation of adults, there is a paucity of literature supporting its use in children. Agitation in Children Agitation in Pregnancy Agitation in Older Patients Patients 65 years or older are particularly susceptible to adverse drug reactions because of coexisting medical illness, use of multiple prescription medications (which increase the risk for drug-drug interactions), and age-associated changes in pharmacokinetics and pharmacodynamics. Research suggests that conventional antipsychotic medications such as haloperidol and droperidol are safe and effective for both psychotic symptoms and nonpsychotic agitated behavior. Significant infection after barb removal is rare, and prophylactic antibiotics are unnecessary. A barb embedded in a vascular structure can probably be removed with manual traction followed by direct pressure on the wound because the size of the barb is similar to the size of devices used to obtain central venous access. Severe involuntary muscle contractions from the electrical discharge has been implicated as a cause of acute thoracic compression fractures. Note: the groove in the shaft (arrow) lines up with the barb tip to aid in removal. Place one hand on the skin surrounding the barb to hold the skin taut and use the other hand to apply direct pressure to the barb. Place one hand on the skin surrounding the barb to hold the skin taut and use the other hand to apply direct pressure to the barb108,109. If the patient cannot tolerate the procedure, inject a small amount of local anesthetic near the barb and use a No. Whenever possible, the least restrictive methods should be used to de-escalate aggressive behavior, or to calm agitated or disruptive individuals, such as a quiet and low-stimulation environment, reasonable bargaining, redirection of the patient, involvement of family, reality orientation, talk down, or a show of force. It is a gray area, indeed, as to when, or to what extent, any intervention is considered necessary to restrain a patient, or to protect a patient or medical personnel from harm. Effective measures are usually initiated by the emergency physician because psychiatric evaluation on such short notice is impractical or unavailable and important decisions must be made immediately with limited data. In that study there was a reduction in personnel injury rates and the contention that one suicide was averted. Standards for restraint and seclusion: Joint Commission on Accreditation of Healthcare Organizations. Fassler D, Cotton N: A national survey on the use of seclusion in the psychiatric treatment of children. Zun lS: A prospective study of the complication rate of use of patient restraint in the emergency department. Joint Commission on Accreditation of Healthcare Organizations: Preventing Restraint Deaths, 1998. A series of 30 cases from the Dade and Broward County Florida Medical Examiner Offices from 1982 to 1990. Ross Dl: An analysis of in-custody deaths and positional asphyxiation, Police Marksman 1996; March/April:1618. Khan A, levy P, DeHorn S, et al: Predictors of mortality in patients with delirium tremens. Sorrentino A: Chemical restraints for the agitated, violent, or psychotic pediatric patient in the emergency department: controversies and recommendations. Thomas H, Jr, Schwartz E, Petrilli R: Droperidol versus haloperidol for chemical restraint of agitated and combative patients. Battaglia J, Moss S, Rush J, et al: Haloperidol, lorazepam, or both for psychotic agitation Breier A, Meehan K, Birkett M, et al: A double-blind, placebo-controlled dose-response comparison of intramuscular olanzapine and haloperidol 1498. Meehan K, Zhang F, David S, et al: A double-blind, randomized comparison of the efficacy and safety of intramuscular injections of olanzapine, lorazepam, or placebo in treating acutely agitated patients diagnosed with bipolar mania. Spina E, de leon J: Metabolic drug interactions with newer antipsychotics: a comparative review. Reich Dl, Silvay G: Ketamine: an update on the first twenty-five years of clinical experience. Bourgoin A, Albanese J, Wereszczynski N, et al: Safety of sedation with ketamine in severe head injury patients: comparison with sufentanil. Melamed E, Oron Y, Ben-Avraham R, et al: the combative multitrauma patient: a protocol for prehospital management. Alexander J, Tharyan P, Adams C, et al: Rapid tranquillisation of violent or agitated patients in a psychiatric emergency setting. Martel M, Sterzinger A, Miner J, et al: Management of acute undifferentiated agitation in the emergency department: a randomized double-blind trial of droperidol, ziprasidone, and midazolam. Zimbroff Dl: Pharmacological control of acute agitation: focus on intramuscular preparations. Brook S: Intramuscular ziprasidone: moving beyond the conventional in the treatment of acute agitation in schizophrenia. Kaloostian P, Tran H: Intracranial taser dart penetration: literature review and surgical management. In addition to medical management, troubleshoot the device and remove it in cases of uncertainty or emergency. To remove the catheter, simply peel off the adhesive and embedded catheter together to discontinue the flow of injected medication into the patient. There are a variety of proprietary pump manufacturers, each with their own device programming. Peabody n addition to cardiac pacemakers and defibrillators, a number of noncardiac devices have been developed for electronic neuromodulation and drug delivery. By 2007, there were more than 375,000 external insulin infusion pumps in use in the United States. Ziconotide is a non-opioid calcium channel blocker, but its use may be complicated by confusion, somnolence, and other neurologic side effects. Although limited data demonstrate efficacy of intrathecal morphine and ziconotide in relieving refractory pain69 and efficacy of intrathecal baclofen in reducing spinal cord injury-induced spasticity,10 more quality clinical evidence is needed. Other medications used include bupivacaine, hydromorphone, fentanyl, sufentanil, clonidine, midazolam, and meperidine. There is minimal data comparing various medications and current regimens have been empirically derived. For intrathecal morphine infusions, approximately 1% of the total daily morphine dose is a standard starting point. Such small intrathecal doses reduce systemic concentrations and minimize side effects. The implanted catheter site varies, but it is commonly placed in the subcutaneous tissue of the abdomen in adults and the buttocks in young children. Similar findings were reported in a 2014 prospective study, in which the most clinically significant adverse events related to insulin infusion devices were hyperglycemia and ketosis. In 29 deaths, problems with the device that were identified included overinfusion, bent cannulas, disconnection, pump alarming, failure to deliver, suspected electromagnetic interference, and display failure. To discontinue the flow of insulin into the patient, simply peel off the adhesive and embedded catheter together. B, the pump is usually implanted in a subcutaneous pocket in a lower abdominal quadrant, with the catheter tunneled subcutaneously to an appropriate lumbar interspace.
Griseofulvin 250 mg purchase amex. Medical Information : Home Remedies to Treat Yeast Infections.
In recent years imperfect fungi definition biology generic griseofulvin 250 mg free shipping, transplantation has been undertaken prior to the development of nodules in order to prevent carcinoma [87 89] fungus gnats cannabis hydroponics purchase griseofulvin overnight delivery. Survival rates at 36 months following transplantation of the liver for this disease have been as high as 87 percent [89] antifungal foot cream 250 mg griseofulvin fast delivery. Tyrosyl compounds in the urine decreased to normal antifungal diy trusted 250 mg griseofulvin, while succinylacetone decreased antifungal infection cream order griseofulvin on line amex, but as far as normal in only one patient [90]. The management of a patient with multiple hypodense hepatic nodules has become complex because of a report that nodules might represent nodular cirrhosis and fatty change [91], and a report that with medical treatment such lesions disappeared [92]. Renal tubular reabsorption of phosphate and bicarbonate may become normal within five days of transplantation; glycosuria and aminoaciduria correct within two weeks [93]. Treatment with 1 mg/kg of this compound has led regularly to improvement in hepatic and renal function, and no side effects have been observed. Concentrations of succinylacetone and -fetoprotein have decreased, and hepatic morphology has improved. Excretion of -aminolevulinic acid has decreased to near normal, and erythrocyte porphobilinogen synthesis increased. This appears to eliminate the neurologic crises of the disease in those properly treated [95]. As of 2003, 369 patients had been treated [96], and treatment was continuing on 293. Withdrawals were 76, of whom 26 had died; 21 had liver failure, of which 12 died; 25 had developed hepatocellular carcinoma, of whom seven had died; and 54 had been transplanted, of whom eight died. Now, approximately 90 percent of those diagnosed before two years of age are alive, some as long as 12 years. There have been only three hepatic cancers in those treated before two years of age, and one of these was present at diagnosis, before treatment. All had rickets and a renal Fanconi syndrome with lowered plasma phosphate prior to treatment. In those with hepatic failure, the recommended dose is 2 mg/kg, allowing the dose to fall with growth to 1 mg/kg before increasing it. A diet low in tyrosine and phenylalanine is also necessary to avoid pathologic elevations of tyrosine. Monitoring of levels of succinylacetone is important to ensure against lapses of compliance with medications. Assay of fumarylacetoacetate fumarylhydrolase in human liver-deficient activity in a case of hereditary tyrosinemia. Acute hereditary tyrosinaemia type I: clinical biochemical and haematological studies in twins. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient. A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxylphenylpyruvate dioxygenase. Lifetime expectancy with dietary treatment in tyrosinemia type I: consequences for timing of liver transplantation. Diagnosis and treatment: some diseases, syndromes, and conditions associated with an unusual odor. Hypermethioninemia: a metabolic disorder associated with cirrhosis islet cell hyperplasia and renal tubular degeneration. Familial cirrhosis of the liver renal tubular defects with rickets and impaired tyrosine metabolism. In: Anonymous Société Belge de Demographic Historiens et Populations: Liber Amicorum Etienne Hélin. The pre- and post-natal diagnosis of tyrosinemia type I and the detection of the carrier state by assay of fumarylacetoacetase. Prenatal diagnosis of hereditary tyrosinaemia type 1 by determination of fumarylacetoacetase in chorionic villus material. Prenatal diagnosis of hereditary tyrosinemia by determination of fumarylacetoacetase in cultured amniotic fluid cells. Prenatal diagnosis of hereditary tyrosinemia: measurement of succinylacetone in amniotic fluid. Stable isotope dilution analysis of succinylacetone using electron capture negative ion mass fragmentography: an accurate approach to the preand neonatal diagnosis of hereditary tyrosinemia type I. Characterization of the human fumarylacetoacetate hydrolase gene and identification of a missense mutation abolishing enzymatic activity. Strong association of hereditary tyrosinemia type 1 with haplotype 6 in FrenchCanadians. Mutations of the fumarylacetoacetate hydrolase gene in four patients with tyrosinemia type I. Geographical and ethnic distribution of mutations of the fumarylacetoacetate hydrolase gene in hereditary tyrosinemia type 1. Pitfalls in the initial diagnosis of tyrosinemia: three case reports and a review of the literature. Tyrosinemia with acute intermittent porphyria: -aminolevulinic acid dehydratase deficiency related to elevated urinary aminolevulinic acid levels. Cardiomyopathy in fumarylacetoacetase deficiency (hereditary tyrosinaemia): a new feature of the disease. Genetic epidemiology of hereditary tyrosinemia in Quebec and in SaguenayLac-StJean. Urinary excretion of succinylacetone and -aminolevulinic acid in patients with hereditary tyrosinemia. Impairment of heme synthesis by succinylacetone: a powerful inhibitor of -aminolevulinate dehydratase activity produced in tyrosinemia. Quantitative determination of succinylacetone in dried blood spots for newborn screening of tyrosinemia type I. Combined newborn screening for succinylacetone, amino acids, and acylcarnitines in dried blood spots. On the renal tubular damage in hereditary tyrosinemia and on the formation of succinylacetoacetate and succinylacetone. Biochemical studies of a patient with hereditary hepatorenal tyrosinemia: evidence of glutathione deficiency. Mechanism of the interaction of coenzyme glutathione and substrate maleylacetone in the presence and absence of enzyme. Tyrosinaemia type I: de novo mutation in liver tissue suppressing an inborn splicing defect. Homotransplantation of the liver in a patient with hepatoma and hereditary tyrosinemia. Multiple hepatic lesions in a girl with tyrosinemia: not always hepatocellular carcinoma. Rapid improvement in the renal tubular dysfunction associated with tyrosinemia following hepatic replacement. Treatment of hereditary tyrosinemia type I by inhibition of 4-hydroxyl-phenylpyruvate dioxygenase. Peripheral neuropathy as the presenting feature of tyrosinemia type I and effectively treated with an inhibitor of 4-hydroxylphenylpyruvate dioxygenase. A majority of patients has the classic phenotype in which life-threatening illness begins in the early days of life, and most patients die if not maintained by the use of mechanical ventilation. Survivors usually display little cognitive development and often have virtually continuous seizures. Enzyme analysis is not generally available; the enzyme is fully expressed only in the liver and brain. The T-protein transfers the -carbon as a methyl group from the H-protein onto tetrahydrofolate yielding methylene tetrahydrofolate with release of ammonia. In patients with nonketotic hyperglycinemia in whom the individual components have been studied, the majority has had defects in the P-protein. Defective activity of the T-protein has also been described, and variant nonketotic hyperglycinemia is caused by defects in the lipoylation of the H-protein or the biosynthesis of the iron sulfur clusters required for lipoate synthesis [5, 6]. Then, usually after the initiation of protein-containing feedings, lethargy develops, along with anorexia and failure to feed or later to suck. Some are ventilated artificially using a respirator for long enough to permit the diagnosis. Subsequent treatment with exchange transfusion, peritoneal dialysis, or sodium benzoate may lead to the initiation of spontaneous respirations and the discontinuation of the respirator which also occurs spontaneously between 10 and 22 days of age. However, there is seldom much evidence of cerebral development and many patients die within the first year of life. The disorder is diagnosed in increasing fashion in neonatal intensive care units of major medical centers, but it is likely that as many or more die neonatal deaths without benefit of diagnosis. They are prominent in almost all patients and may be virtually continuous [1, 17, 19]. Hiccupping is common and often persistent [11] and, with some frequency, we have obtained historical evidence of recurrent prenatal hiccupping. The typical pattern of burst-suppression is one of periodic or pseudoperiodic areas of large-amplitude sharp waves on a low voltage background [21, 22]. The burst-suppression pattern has been observed as early as 30 minutes after birth [22]. This pattern, typical of the neonate, may change to hypsarrhythmia in later infancy. Patients surviving the acute neonatal crisis develop a pattern of cerebral palsy with spasticity and hypertonia [1, 3], though they may be hypotonic throughout infancy [3, 11]. The patient may be completely unaware of surroundings and have few spontaneous movements. Following exchange transfusion, assisted ventilation could be discontinued, but the patient was still in the intensive care unit and unresponsive. He survived a neonatal requirement for assisted ventilation following treatment with sodium benzoate but had little development or awareness of his environment. Gastroesophageal reflux develops frequently, and gavage or gastrostomy feeding may be required. The natural history of the disease was recorded through two clinical surveys covering close to 150 patients [24, 25]. Nonketotic hyperglycinemia is heterogeneous and, while the majority of patients display the classic phenotype, a small number has been reported in whom a variety of milder or attenuated forms have been observed. Children with classical nonketotic hyperglycinemia often succumb early with a median age of death of 31. More girls than boys died in the newborn period; mean age of female death was less than one month, while that of boys was 2. At the extreme from the classic, three affected girls [26] had only mild impairment; only one of the three was in an institution. Other families have been reported in which there was mild developmental delay [2732]. Acute febrile illness has been associated with involuntary movements, paresis of upward gaze, and delirium. Severe mental impairment and seizures may be found despite an atypical late onset [32]. A milder course manifests with a better recovery in the first two months of life with increased wakefulness and no development of spasticity in infancy. There are few or no seizures, usually controlled by benzoate therapy and/or by a single anticonvulsant medication. More prominent than seizures are hyperactivity, behavioral problems, chorea and intermittent episodes of lethargy and ataxia. Most of these patients make developmental progress but remain mentally retarded, with developmental quotients varying between 20 and 70. Symptoms, in addition to a presentation like classical nonketotic hyperglycinemia and/or a mitochondrial disorder, can be leukoencephalopathy, optic atrophy, cardiomyopathy, pulmonary hypertension, lactic acidosis, and regression. We encountered a boy with muscular hypotonia, mild developmental delay and deceleration of head growth at the age of 17 months when attacks of recurrent vomiting started. From the age of 19 months, he rapidly deteriorated in crises with disturbed consciousness usually triggered by intercurrent illnesses. This is the sulfur donor in the synthesis of lipoic acid, which becomes covalently attached to specific lysine residues of mitochondrial enzymes, among them the H protein of the glycine cleavage system. We have again encountered such different presentations [35] as a neurodegenerative disease not unlike Tay-Sachs or Krabbe disease. The patient developed relatively normally for the first months of life and then, in the second half of the first year, showed progressive cerebral deterioration. The corpus callosum was abnormally thin in all patients and volume loss was both supra- and infratentorial. T 2-weighted images revealed decreased or absent myelination in the supratentorial white. These observations are consistent with reported neuropathology, including atrophy and corpus callosal thinning. Normal cognitive and neurologic function at follow up has been reported but, in the patients followed by us, there was mild mental retardation at school age. In these patients, the glycine cleavage enzyme is not primarily affected and mutations are not found in the specific genes. In the initial phase, the lack of the pattern of lesions on diffusion-weighted brain images typical for nonketotic hyperglycinemia can give some help in the differential diagnosis. Defective activity of overall glycine cleavage was first described in vivo in studies of the metabolism of 14C-labeled glycine [2]. Assay of the enzyme in the liver homogenates was reported in 1969 by Tada and colleagues [43]. The enzyme system is expressed in the liver, kidney, and brain, and until recently, could only be demonstrated in those tissues. Later, it has been shown that the system is induced in the transformation of B lymphocytes with EpsteinBarr virus [44].
References
- Altmann-Schneider I, Trompet S, de Craen AJ, et al. Cerebral microbleeds are predictive of mortality in the elderly. Stroke 2011;42:638.
- Weyman AE. Assessment of mitral stenosis: role of real-time 3D TEE. ]ACC Cardiovasc Imaging. 2011;4(6):589-591.
- Ferrari, G. C., Miranda, A., Sansonna, F., et al. Laparoscopic repair of incisional hernias located on the abdominal borders: a retrospective critical review. Surg Laparosc Endosc Percutan Tech. 2009; 19:348-352.
- Hall J, Hammerich K, Roberts P. New paradigms in the management of diverticular disease. Curr Probl Surg. 2010;47(9):680-735.
- Heilman KM, Bowers D, Watson RT. Pseudoneglect in a patient with partial callosal disconnection. Brain 1984;107:519.
- Esen S, Akar?rmak U, Ayd?n FY, Unalan H. Clinical evaluation during the acute exacerbation of knee osteoarthritis: the impact of diagnostic ultrasonography. Rheumatol Int 2013; 33(3):711-7.
- Seligsohn U. High frequency of factor XI (PTA) deficiency in Ashkenazi Jews. Blood 51:1223, 1978.