

Ezetimibe
| Contato
Página Inicial
Jonathan Abrams, MD
- Distinguished Professor of Medicine
- University of New Mexico
- Cardiology Division
- Albuquerque, New Mexico
A fairly close and direct relationship exists between axon diameter and internodal length cholesterol test malaysia ezetimibe 10 mg buy low cost, small fibres having short internodes and large fibres longer internodes cholesterol what is it ezetimibe 10 mg purchase visa. During development cholesterol levels by age ezetimibe 10 mg fast delivery, the total number of Schwann cells along the length of an axon is fixed cholesterol chart american heart association discount 10 mg ezetimibe, and subsequent growth in fibre length is accommodated by elongation of this finite population of Schwann cells and their contained myelin segments cholesterol vegan diet buy genuine ezetimibe. Internode length can be evaluated diagnostically if individual fibres are teased out and examined longitudinally. Excessively short myelin segments occur, representing a manifestation of previous Schwann cell damage with subsequent regeneration. Histochemical studies at a fine structural level have demonstrated that the gap substance filling the space between the Schwann cell nodal processes and the basal lamina contains glycosaminoglycans with cationbinding and -exchange properties. The gap substance may act as an ion-exchange buffer with a capacity to concentrate and maintain a high but osmotically inactive concentration of sodium ions immediately adjacent to the axolemma, and to limit diffusion away from the axolemma of potassium ions, which have passed outwards through the membrane during the passage of an action potential. The axolemma underlying the nodal region is rich in sodium channels important for impulse propagation (see Transmission of the Nerve Impulse, p. In contrast, the normal internodal sodium channel concentration is insufficient to maintain a propagated impulse. Both the arrangement and the content of organelles within the nodal and juxtanodal axon show consistent differences from those seen elsewhere along the internode. BasIc PathologIcal MechanIsMs Although there are several hundred experimental, veterinary and clinical conditions that result in dysfunction of peripheral nerves, their pathological substrates can be classified into a few patterns of injury. These important processes can be divided into axonal degeneration, segmental demyelination, secondary demyelination, axonal atrophy and axonal dystrophy, each of which is discussed subsequently in this section. In a number of conditions, several of these patterns occur simultaneously or sequentially, for example sublethal degenerative axonal changes or axonal atrophy resulting in secondary demyelination. The structural and molecular events associated with axonal injury have been studied most extensively in nerve damaged by mechanical crush injury, i. Axonal Events Although the ability to conduct impulses is maintained for as long as 48 hours distal to a site of axonal transection,61 a variety of structural and biochemical changes begins almost immediately. Disorganization of axonal microtubules and the endoplasmic reticulum occurs within 1224 hours. Relatively few neurofilaments are evident in transverse sections of the nodal axon, and these retain the same packing density as in the internodal regions. Mitochondria are approximately five times more numerous in the paranode, reflecting the existence of continuous energy-consuming activity in this region of the Schwann cell. Schwann cell mitochondria help maintain the axoglial interactions required for the long-term support of Basic Pathological Mechanisms 1425 Axon Axonal crush Schwann cell Basal lamina 24 Macrophage Proliferated Schwann cells Regeneration 24. Dilated expansions of unmyelinated axons close to the site of transection are filled with numerous subcellular organelles. The axon shows granular disintegration of its cytoskeleton (microtubules and neurofilaments) in the presence of an intact myelin sheath. Similar mechanisms promote axonal degeneration after exposure to chemotherapy, evidence for participation in a general axon self-destruction program. Differences between protection of sensory and motor axons and their endings in Wlds mice may be accounted for by variation in the overall amount of expression of Wlds protein expressed in sensory and motor neurones and their axonal branches. Accumulation of mitochondrial pathology, possibly reflecting injury secondary to local oxidative stress to the mitochondrial genome or difficulty with mitochondrial calcium homeostasis, may underlie distal axonopathy. Raff and colleagues proposed that dying-back axonal degeneration results from the activation of a self-destruct programme in the distal parts of an axon in response to a neuronal insult. Initial digestion of the axon and its myelin sheath occurs within the Schwann cell: (a) 1-m plastic section, ×600; (b) electron micrograph, ×7500. Schwann cell processes are held within the corrugated remnants of the original basal membrane (arrow) that enclosed the Schwann cellaxon unit. Schwann cell proliferation is greater in nerves with large myelinated axons than in those containing mostly small fibres. These changes involve the axonal growth cone and Schwann cell-derived neuregulin-1, functioning as a dedifferentiation factor, and upregulation of Schwann cell erbB2 and erbB3 neuregulin receptors, 37 days postaxotomy. Schwann cells not associated with axons will, in time, undergo apoptosis, whereas those in contact with axons will proliferate and migrate along the axon as needed, not synthesizing myelin until a proper numerical relationship is established. Foamy macrophages (arrows), swollen with partially degraded axonal and myelin debris, accumulate near an endoneurial venule during axonal degeneration. Macrophages also participate in the synthesis and secretion of apolipoprotein E for reutilizing lipids. Lipid may accumulate in macrophages, fibroblasts, Schwann and perineurial cells and endothelial cells, and this material may persist for months. It can be difficult to distinguish reinnervated bands of Büngner from Schwann processes alone, although this is often simplified by the demonstration of significant aggregates of ribosomes in Schwann cell processes, increased Schwann cell cytoplasmic osmiophilia, prominence of cytoplasmic filaments in Schwann cells, and microtubules in axonal processes. Depletion of Schwann cells does not influence axonal elongation when the basal lamina remains in continuity, which suggests a critical role of extracellular matrix proteins in axon regeneration. In experimental and clinical crush injuries, a wave of axonal degeneration may precede regeneration; however, in most clinical cases, axonal degenerative and regenerative processes may occur simultaneously. Axonal regeneration is partially dependent on the reutilization of cholesterol within the endoneurium. One dominant axon emerges and the others regress, leaving behind a myelinated axon Basic Pathological Mechanisms (a) (b) 1429 24 24. A macrophage (arrow) is shown that has finished removing axonal and myelin debris from one Schwann cell containing regenerating axonal sprouts and is sending a process to a second Schwann cell at the upper margin of the micrograph. Failure of regeneration/reconnection results in loss of axon diameter proximal to the injury site61 and relatively thick myelin for axon calibre. Large clusters of frustrated regenerating axons composing a traumatic neuroma suggest the failure to eliminate supernumerary axons. Regenerating unmyelinated axons have a more loose arrangement of axonal sprouts with basal lamina and Schwann cell processes and may persist as groups not associated with Schwann cell processes. The mechanisms by which regenerating axons result in precise matching of the original axon with its Schwann cell tube are unknown. Experimental freeze injury, in which regenerating axons are typically contained by their original basal lamina, favours regeneration. Regenerating rat femoral motor axons send out collateral branches into both appropriate and inappropriate nerve branches, the latter of which are pruned, perhaps as the result of deficient neurotrophic signals. A chronic severe neuropathic process may result in an end-stage nerve in which only rare preserved axons are accompanied by scattered fibroblasts and diminished 1430 (a) Chapter 24 Diseases of Peripheral Nerves (b) 24. Although there are similarities between the biochemical and pathological processes occurring in axonal degeneration and wallerian degeneration, in the latter the histological changes appear stereotyped and synchronized. The histological appearance of wallerian degeneration is also stereotyped in contrast to axonal degenerations, in which distinctive pathological signatures differ between specific axonal degenerations. Pathological signatures include neurofilamentous aggregates (acrylamide, hexacarbon, 24. A growth cone (arrow) containing large numbers of tubulovesicular elements is enveloped by Schwann cell processes. Teased fibre preparations may contribute to understanding the pathological history of individual axons in which axonal degeneration has previously occurred. The most definitive proof of axonal atrophy requires morphometric determinations in which the plot of the ratio of axonal diameter to axon 1 myelin thickness. There are normal variations in axon diameter and myelin thickness near the node of Ranvier. Groups of small thinly myelinated axons (red arrows), which represent maturing sprouts from a single parent axon, are visible by light microscopy (a) and ultrastructure (b). Occasional regenerated axons have myelin sheaths too thin for axon calibre (blue arrows). All visible myelinated fibres are degenerating synchronously in this patient with traumatic avulsion of the brachial plexus. Dystrophic segments may not always represent termini and have been demonstrated as multifocal expansions along individual axons. The continued supply of membranous elements may result in complex, occasionally crystalline membranous aggregates. Studies of gad (gracile axonal dystrophy) mutant mice identified an inactivation of the 24. The axon of this myelinated axon in a solvent-abusing patient is shrunken and atrophic and is enclosed by a collapsed and folded myelin sheath. Initial myelinopathy may be subtle, with vesicular degeneration, splitting of myelin lamellae, widening of the nodal gap or unusual folding of the myelin sheath. Treatment of Schwann cell cultures with the calcium ionophore A23187 increases Ca2+ leakage into Schwann cell cytoplasm, activates endogenous phospholipase A2 and results in demyelination. If sustained or of appropriate severity, a pathological process involving the Schwann cell or the myelin sheath results in the degeneration of the myelin sheath and, if the original Schwann cell survives 24. An unmyelinated axon in a diabetic sympathetic ganglion is swollen by unusual membranous collections. Remyelination the insult, myelin degradation typically begins within the Schwann cell cytoplasm. As in axonal degeneration, haematogenous and intrinsic macrophages engulf the debris provided by the Schwann cell. Myelin debris is usually cleared rapidly, and the presence of macrophages containing this debris after several weeks is evidence for ongoing demyelination. Schwann cell proliferation results in the eventual remyelination of individual internodes, typically replacing each lost myelin internode with several shorter intercalated internodes. Remyelination requires Schwann cell proliferation from demyelinated segments as well as adjacent unmyelinated axons. The presence of onion bulbs suggests a cyclical demyelinative/remyelinative process active for several months or more. Although experimental insults may produce acute demyelination followed by remyelination, in nearly all demyelinative neuropathies, demyelination and remyelination occur concurrently. Teased fibre analysis is particularly useful for establishing that a neuropathic process has a demyelinating component. Plotting axonal diameter versus internodal length in teased fibre preparations shows marked variation from node to node in previously demyelinated and remyelinated nerves compared with normal nerves; this pattern corresponds to the replacement of myelin originally lost from the domain of a single Schwann cell by several proliferated Schwann cells with smaller territories. In contrast to segmental demyelination, in which foci of demyelination are located randomly on individual axons, secondary demyelination preferentially involves selected axons, which may show atrophic or other degenerative axonopathic changes, while entirely sparing other axons. The mechanism appears to involve an abnormality in the relationship of the Schwann cell to its ensheathed axon, resulting sequentially in axonal atrophy and myelin wrinkling, followed by paranodal demyelination and segmental demyelination, and remyelination that may be cyclical. The presence of onion bulbs is most typically associated with primary segmental demyelination rather than secondary demyelination. This compressed axon is surrounded by markedly thickened redundant loops of myelin. Primary inherited peripheral neuropathies are the most common monogenetically inherited diseases of the nervous system, with a prevalence between 1 and 4 per 10 000. Repeated episodes of primary segmental demyelination and remyelination lead to the formation of onion bulbs, a term that refers to imbricated layers of supernumerary Schwann cell processes arranged in rings around the long axis of nerve fibres separated by collagen fibres. Patients with pure motor involvement can be separated as examples of distal muscular atrophy. Patients develop weakness and wasting distally in the lower extremities, associated with areflexia and sensory loss. Pes cavus, hammer toes and scoliosis may be present as early manifestations from childhood. Visible and palpable hypertrophy of peripheral nerves, especially the greater auricular nerve, can be found. Histology reveals expanded fascicular areas because of increased matrix and collagen; there are reduced numbers of myelinated nerve fibres, with a deficiency of both large- and small-diameter elements and evidence of extensive segmental demyelination and remyelination. Mfn2 loss of function inhibits pyruvate, glucose and fatty acid Inherited Neuropathy 1437 oxidation and reduces mitochondrial membrane potential. Histologically, this disorder displays ventral horn and spinal ganglia neuronal loss, maximal in the lumbosacral region, and tract degeneration in the gracilis fasciculi. The onset of this form of the disease is in the second decade, with rapid progression involving upper limbs and proximal muscles, leading to severe disability. Biopsies reveal a reduction of myelinated axons and clusters of regenerating units. This suggests the existence of distinct functional domains in lamin A/C that are essential for different cell types. As is typical for genetic processes, the histopathological picture appears chronic, with no active 24. The mode of inheritance can be autosomal dominant or recessive, but many cases are sporadic. In some patients with a seemingly recessive mode of inheritance, dominant de novo mutations have been identified. The clinical features include sensory motor neuropathy with an aggressive course, ataxia, skeletal abnormalities and markedly enlarged nerves. Nerve biopsy shows marked reduction or even absence of myelin and onion bulbs composed of basal lamina reduplication. A subset of patients with congenital amyelination manifest a most severe syndrome exhibiting flaccidity at birth, often with arthrogryposis, respiratory distress and swallowing difficulties, leading to death within days or months. The gait deteriorates, and the patient becomes wheelchair-bound, with mild sensory and hearing loss. Nerve biopsy shows features of severe reduction of myelinated fibre density and hypomyelination. Onset of symptoms is in the second or third year of life, with a relatively severe neuropathy, both distal and proximal, followed by a relentlessly progressive course culminating in wheelchair dependency and early death. Nerve biopsy shows loss of large myelinated fibres; few fibres have a diameter larger than 8 m. This rare recessive demyelinating syndrome also includes hearing loss and dysmorphism and affects individuals of the Romany population settled near the town of Lom in Bulgaria.
Loss of papillary architecture cholesterol lowering diet plan australia purchase generic ezetimibe on line, cytological pleomorphism and abundant mitotic figures characterize this tumour cholesterol score of 6.3 discount ezetimibe 10 mg with amex. Papillomas are consistently positive for pancytokeratin and vimentin normal cholesterol levels nz generic ezetimibe 10 mg with mastercard, with pancytokeratin often producing a diffuse or dot-like cytoplasmic pattern of reactivity cholesterol lowering foods eggs order ezetimibe with visa. S-100 immunoreactivity is greater in fourth ventricular tumours compared to those of the lateral ventricle cholesterol ratio below 3 cheap ezetimibe 10 mg without a prescription. In adults, other intraventricular tumours include subependymomas, subependymal giant cell astrocytomas, meningiomas, ependymomas or metastases. The majority of these can be easily differentiated from choroid plexus tumours based on simple histologic findings, with a few exceptions as detailed later. Differential diagnostic considerations encompass several histopathologically similar primary and secondary tumours with papillary architecture, as well as normal choroid plexus. The distinction between papilloma and normal/ hyperplastic choroid plexus can be difficult, particularly in the setting of a tiny biopsy. However, papillomas typically lose the characteristic cobblestone-like surface of normal choroid plexus with the latter showing partial spaces between surface epithelial cells. Evaluation of mitotic activity, epithelial architecture and cellular atypia, along with careful consideration of the clinical context, can also help distinguish papillomas from reactive proliferations. These neoplasms are most often considered in the differential diagnosis in children, whereas metastatic papillary carcinoma must be excluded in adults with suspected choroid plexus carcinoma. Similar to choroid plexus papillomas, papillary ependymoma and astroblastoma all feature papillary architecture, but differ in their immunoprofiles. Additionally, E-cadherin positivity and absence of nerve cell adhesion molecule staining supports a diagnosis of choroid plexus carcinoma. Although lacking absolute specificity, transthyretin and S-100 positivity supports choroid plexus carcinoma. As a general rule, few metastatic papillary carcinomas appear as well differentiated as choroid plexus papillomas, and choroid plexus carcinomas are so rare in adults that metastatic papillary carcinoma must be the presumptive diagnosis until proven otherwise. Interactions of viral proteins with p53 and pRb have been identified; however, the biological significance of these interactions has not been fully elucidated. In multiple studies of populations with a high prevalence of LiFraumeni syndrome, an increased ratio of carcinomas to papillomas (1:0. Although recurrences are unusual, papillomas may recur many years after initial presentation, particularly in the context of subtotal removal. Chemotherapy is usually employed as adjuvant treatment for carcinomas, and radiation may be appropriate for adults and older children. Craniospinal irradiation may be helpful in those cases with subtotal resection and/ or leptomeningeal dissemination. Molecular genetIcs Classic and molecular cytogenetic assessments of choroid plexus tumours show that chromosomal imbalance is common in both papillomas and carcinomas. Choroid plexus papillomas are typically hyperdiploid, with gains identified on chromosomes 5, 7, 9, 12, 15, 17, 18, 20 and 21, and losses on chromosomes 10 and 22q. These differences in genetic abnormalities support the theory that choroid plexus tumours of different grade develop de novo, with malignant transformation from papilloma to carcinoma seldom occurring. Fetal tumors of the choroid plexus: is differential diagnosis between papilloma 4. Papillomas and carcinomas of the choroid plexus: histological and immunohistochemical studies and comparison with normal fetal choroid plexus. Choroid plexus tumors differ from metastatic carcinomas by expression of the excitatory amino acid transporter-1. Villous hypertrophy versus choroid plexus papilloma: a case report demonstrating a diagnostic role for the proliferation index. Comparative genomic hybridization detects specific cytogenetic abnormalities in pediatric ependymomas and choroid plexus papillomas. Surgical removal of bilateral papillomas of the choroid plexus of the lateral ventricles with resolution of hydrocephalus. Cytokeratin 7 and 20 expression in choroid plexus tumors: utility in differentiating these neoplasms from metastatic carcinomas. Prognostic implications of atypical histologic features in choroid plexus papilloma. Diffuse craniospinal seeding from a benign fourth ventricle choroid plexus papilloma. Purely cystic form of choroid plexus papilloma with acute hydrocephalus in an infant. Clinicopathologic correlations in epithelial choroid plexus neoplasms: a study of 52 cases. Atypical teratoid/rhabdoid tumors may show morphological and immunohistochemical features seen in choroid plexus tumors. Congenital hydrocephalus due to villous hypertrophy of the telencephalic choroid plexuses. Magnetic resonance imaging in the diagnosis and management of choroid plexus carcinoma in children. Rhabdoid choroid plexus carcinoma: a rare 31 31 Chapter Other Glial Neoplasms Daniel J Brat Introduction. These entities are separated from the more common astrocytic, oligodendroglial and ependymal tumour categories because their undefined histogenesis precludes a definite assignment to any other group. Because of its rarity in pure form and the fact that focal astroblastic features may be encountered in otherwise typical astrocytic and ependymal tumours, it is not entirely clear whether astroblastoma should be regarded as a distinct tumour entity. However, based on several well-defined patient series and a greater emphasis on definitive diagnostic elements, astroblastoma is now generally regarded as a unique type of glial neoplasm with distinctive clinical, radiological, pathological and genetic features. Rare examples have been reported in the cerebellum, brain stem, optic nerves and cauda equina. Anaplastic forms may demonstrate greater peritumoural oedema as a result of their rapid growth. Taken together, the radiological features may help distinguish astroblastomas from ependymomas and diffusely infiltrating gliomas. Areas of necrosis may be discernible, particularly in large lesions and anaplastic variants. Incidence, Age and sex distribution Astroblastomas are rare and epidemiological data are imprecise. The tumours commonly affect adolescents and young adults, with most cases presenting in the first three decades. In this instance, in which the tumour was well differentiated, there is only slight peritumoural oedema with partial compression of the adjacent ventricle. Chordoid Glioma of the Third Ventricle 1719 that radiate towards a central vessel. In tissue sections and on smear preparations, there is a paucity of glial fibrillarity. Other areas often lack astroblastic pseudorosettes, consisting of solid sheets of moderately pleomorphic epithelioid cells with eccentric round to oval nuclei. In contrast to ependymomas, astroblastomas typically arise superficially in the cerebral hemispheres. The presence of classic ependymal pseudorosettes, as well as true ependymal canals and rosettes, helps to distinguish ependymoma. Cytologically, astroblastomas appear more epithelioid and lack the thin fibrillar processes of ependymoma. In addition, perivascular hyalinisation is usually more prominent in astroblastomas than in ependymomas. The angiocentric glioma combines the histopathological features of a diffuse astrocytoma and ependymoma, and may focally resemble an astroblastoma. However, the predominant wrapping around small blood vessels is distinct from the astroblastic pseudorosette, whereas the infiltrative growth is more readily apparent. On the other hand, Rubinstein and Herman found ultrastructural characteristics suggesting an origin from tanycytes. Astroblastomas typically lack cytokeratin expression, although there has been one report of positivity for low molecular weight cytokeratins. Taken together, these data suggest that astroblastomas have a cytogenetic profile that differs from more common astrocytic, oligodendroglial and ependymal tumours. However, further studies are needed to substantiate this hypothesis and to identify astroblastoma-specific gene alterations. Studies with well-described histologic criteria revealed a better prognosis for low-grade than high-grade (anaplastic) variants, with higher recurrence rates in the latter. Two tumours displayed intermediate ultrastructural features between those of astrocytes and ependymal cells. Focal perivascular orientation resembling astroblastic pseudorosettes may be seen in otherwise typical diffuse astrocytomas, including glioblastomas. Immunohistochemistry the immunoprofile of chordoid gliomas greatly aids in the diagnosis. Epidermal growth factor receptors and merlin/schwannomin are expressed, whereas nuclear p53 staining is weak or absent. Neuronal and neuroendocrine markers (synaptophysin, neurofilaments, chromogranin A) are consistently negative. Neuroradiologically, chordoid gliomas present with strikingly similar imaging features. Most tumours abut the hypothalamus and some appear to have an intrinsic anterior hypothalamic component, suggesting a potential site of origin. Other studies observed ultrastructural features suggestive of ependymal lineage, including the formation of microvilli and cilia. The most important alternative histological diagnoses to consider are chordoid meningioma and chordoma. In addition, chordomas contain physalipherous cells, which are not seen in chordoid gliomas. Other radiological differential diagnoses are readily distinguished by histopathological evaluation. They typically adhere to the ventricular walls, expand the third ventricle and cause obstructive hydrocephalus. Tumour cells may form coarsely fibrillar processes, embedded in an Alcian blue positive, mucinous and sometimes vacuolated matrix. Other minor architectural patterns include papillary, alveolar and pseudoglandular forms. Histogenesis One of the most striking features of chordoid gliomas is their predilection for the anterior third ventricle and hypothalamus. In keeping with other studies, they found evidence of ependymal differentiation (microvilli and hemidesmosome-like structures). Further evidence of specialized ependymal differentiation has come from a report that demonstrated abnormal cilia in a juxtanuclear location. Borders typically show chronic inflammation and Rosenthal fibres within the neighbouring brain. Tumour cells are uniform, spindled with oval or elongated nuclei and speckled chromatin as well as pink, tapering cytoplasm. These slender cells are most frequently oriented parallel to vessels, sometimes expanding perivascular spaces with streaming arrays of either single or multilayered cells. In some examples, tumour cells are radially oriented to vessels in a pattern, highly reminiscent of ependymal pseudorosettes. In a small subset, a similar tendency to orient perpendicularly to the pia mater is seen, giving a palisading appearance at the brain surface. In addition to the perivascular and subpial distribution, scattered single cells or cell clusters are present at low density within the cortical and subcortical parenchyma. Non-neoplastic elements, including cortical neurons and neuropil, are entrapped within the tumour, consistent with an infiltrative growth pattern. In lesions resected from adults with long-standing histories of seizures, neurofibrillary tangles may be seen in native neurons. Cases with higher indices have been described, yet it is unclear if this has an impact on clinical behaviour. Gross total resection is the treatment of choice and may result in long-term recurrence-free survival. Consequently, several patients have been reported with disabling post-operative neurological complications and/or early postoperative death. Indeed, the majority of deaths recorded for patients with chordoid glioma occur in the first four weeks post-operatively. The infiltrative growth pattern is also highlighted with stains for neurofilament protein, which show entrapped intratumoural axons. Ependymal differentiation is also seen on electron microscopy, which demonstrates microlumen formation, microvilli, cilia, and complex, zipper-like intermediate junctions. Nearly all patients have long-standing epilepsy that is refractory to medical treatment. Tumours are typically centred in the cortex, but often extend into subcortical regions. The distinction can be more problematic when ependymomas occur within the brain parenchyma, such as is seen with cortical ependymoma. Although pilocytic astrocytomas can have a perivascular growth pattern and orient toward vessels, the degree is usually less dramatic than that of angiocentric glioma. Moreover, unlike angiocentric glioma, pilocytic astrocytomas tend to form solid components that push aside, rather than invade adjacent brain. Diffuse astrocytoma shares the infiltrative growth pattern and elongate nuclei seen in angiocentric glioma, but it lacks the angiocentricity, typically involves larger portions of the brain, and shows greater nuclear hyperchromasia and pleomorphism. Despite these ependymal features, however, tumours show an infiltrative growth pattern. Astroblastoma presenting with intracerebral hemorrhage misdiagnosed as dural arteriovenous fistula: review of a rare entity. A classification of tumors of the glioma group on a histogenetic basis with a correlated study of prognosis.
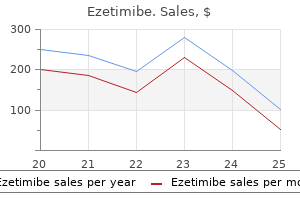
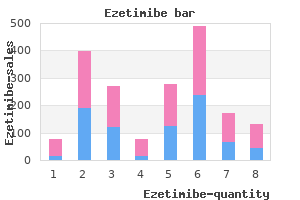
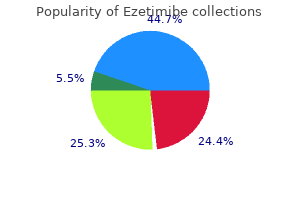
Cheap ezetimibe 10 mg amex. Saturated Fats and Testosterone.
NasuHakola disease: a review of its leukoencephalopathic and membranolipodystrophic features cholesterol test understanding results buy ezetimibe with american express. Ballooned neurons in progressive supranuclear palsy are usually due to concurrent argyrophilic grain disease cholesterol medication for elderly generic ezetimibe 10 mg online. Proposed neuropathological criteria for the post mortem diagnosis of multiple system atrophy cholesterol levels test results order ezetimibe 10 mg with amex. Lewy bodies are not increased in progressive supranuclear palsy compared with normal controls cholesterol test instructions ezetimibe 10 mg order online. Constant involvement of the Betz cells and pyramidal tract in multiple system atrophy: a clinicopathological study of seven autopsy cases cholesterol your hair purchase 10 mg ezetimibe amex. Constant and severe involvement of Betz cells in corticobasal degeneration is not consistent with pyramidal signs: a clinicopathological study of ten autopsy cases. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. A historical analysis of the relationship between encephalitis lethargica and postencephalitic parkinsonism: a complex rather than a direct relationship. Encephalitis lethargica: part of a spectrum of post-streptococcal autoimmune diseases Corticobasal degeneration: etiopathological significance of the cytoskeletal alterations. Multiple system atrophy with severe involvement of the motor cortical areas and cerebral white matter. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Incidence of amyotrophic lateral sclerosis and of the parkinsonism-dementia complex of Guam, 19501989. Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Tau-positive fine granules in the cerebral white matter: a novel finding among the tauopathies exclusive to parkinsonism-dementia complex of Guam. Patterns and stages of alpha-synucleinopathy: relevance in a population-based cohort. Hereditary neuronal intranuclear inclusion disease with autonomic failure and cerebellar degeneration. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Clinicopathological investigation of vascular parkinsonism, including clinical criteria for diagnosis. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Characteristic manifestations of ataxia include postural and gait abnormalities, incoordination of limb movements, dysarthria and oculomotor disturbances. Postural changes are typified by swaying instability of the trunk while standing or even sitting. A wide-based stance is assumed, but patients may still have difficulty maintaining balance. Gait is irregular, wide-based and staggering, with loss of the ability to tandem walk. Limb abnormalities include dysmetria, an inability to perform movements with proper timing and trajectory. Intention tremor, loss of fluidity of movement, inability to control force of movement and dysdiadochokinesia are features of dysmetria. The last of these refers to a reduced ability to perform rapid, alternating movements. Oculomotor changes may consist of disorders of ocular fixation, ocular alignment, visual pursuit, saccadic movements and nystagmus. A guideline for the clinical diagnosis and management of adult ataxias has been published by the European Federation of Neurological Societies. Historically, classification of these conditions has been difficult because neither pathological nor clinical characterizations could reliably describe definitive disorders, particularly genetic forms of ataxia. Advances in the identification of the genetic bases of many of these conditions have resulted in a more rational basis of classification, but many of their clinicopathological features remain complicated by phenotypic heterogeneity. Previous pathological classifications have focused on three major patterns of neurodegeneration: Cerebellar cortical degeneration is characterized by loss of Purkinje cells, with less or negligible involvement of cerebellar granule cells or cortical interneurons. Loss of neurons in the inferior olivary nuclei occurs frequently and is believed to result from retrograde trans-synaptic degeneration. The cranial nerve nuclei, deep cerebellar nuclei, spinal cord, basal ganglia, substantia nigra and red nuclei also may be affected. Spinocerebellar degeneration is characterized by loss of cerebellar afferent projections, with less or negligible involvement of the cerebellar cortex. Involvement of these neuroanatomical sites may be the Unfortunately, marked pathological heterogeneity results in overlap among these three, seemingly distinct, pathological subtypes. Furthermore, many of the pathological descriptions of disease entities were made before discovery of their genetic aetiologies, so that correlation of clinical, pathological and genetic information may be difficult. Despite numerous patterns of regional involvement, the gross and histopathological features in individual areas are stereotypical. Loss of Purkinje cells is typically accompanied by hypertrophy and hyperplasia of Bergmann glia, with shrinkage of the molecular layer, resulting in at least a transiently increased density of stellate and basket neurons. Loss of granular neurons is usually less severe than loss of Purkinje cells, but in longstanding disease there may be a significantly decreased density, often with an inconspicuous glial reaction. The interneurons of the molecular layer may be depleted in later stages of cortical degeneration. The reduction of Purkinje cell axon terminals in the deep cerebellar nuclei may induce gliosis, with little or no obvious neuronal loss, although in some conditions the dentate neurons are a primary target of the disease process. In the latter instance, there is often associated atrophy of the superior cerebellar peduncles. Inferior olivary atrophy usually accompanies chronic Classification of Degenerative Ataxias (a) 801 13 (b) (b) 13. There is cortical atrophy that is most pronounced anterior and superior, with sparing of the tonsil. Note the atrophic myelin pallor in the deep and foliar white matter, due to loss of mossy fibres and Purkinje cell axons, but with sparing of the dentate efferent fibres. Note pallor of middle cerebral peduncles and preservation of superior cerebellar peduncles. In some forms of ataxia, it is likely that olivary atrophy is a primary manifestation of the disease. In more complex degenerations, with brain stem and spinal cord involvement, there are other characteristic features. Degeneration of the basal pontine nuclei is reflected by gross atrophy of the basis pontis, loss of transverse fibres, shrinkage of the middle cerebellar peduncles and loss of volume of the cerebellar white matter. Microscopically, these changes are not unique but show loss of neurons and fibre tracts, with accompanying astrocytosis. Similar histopathological changes can be seen in other brain stem and spinal cord nuclei and tracts. New histopathological diagnostic tools have emerged from increased understanding of many of the degenerative ataxias. Likewise, the glial cytoplasmic inclusions characteristic of multiple system atrophy, a common sporadic form of ataxia, are immunoreactive for synuclein and ubiquitin. Calbindin is a useful marker for Purkinje neurons and transmitter-specific markers have utility in elucidating specific types of neuronal loss. A few other rarer forms of recessive ataxia are discussed later and listed in Table 13. Onset typically occurs early in the second decade of life, but the range extends from 18 months of age to well into the third decade. The first manifestation of disease usually is instability of gait or a sense of clumsiness. Truncal ataxia progresses to limb ataxia, dysarthria, areflexia and sensory neuropathy with distal loss of joint position and vibration sense. As the course progresses, there is lower extremity weakness with extensor plantar reflexes. Distal amyotrophy in the legs and hands and autonomic abnormalities, such as cold cyanotic feet, are common. Oculomotor changes are nearly always present, but nystagmus is relatively infrequent. Most patients have a cardiomyopathy, which is frequently asymptomatic but is usually more severe in patients with early onset. Abnormal glucose tolerance is present in about a quarter of patients, 10 per cent overall having insulin-dependent diabetes mellitus. Death occurs most frequently in the fourth decade of life, but prolonged survival is common, particularly when diabetes and cardiac disease are absent or treated effectively. It encodes an 18-kDa protein, frataxin,13 which is expressed widely, with greatest levels in human heart and spinal cord; however, frataxin is also is present in brain, liver, skeletal muscle and pancreas. The amount of expressed protein correlates inversely with repeat length and is influenced most by the size of the shorter of the two affected alleles. Most point mutations result in complete loss of function of frataxin, but several are associated with a milder phenotype, suggesting some preserved function. Although the normal function of frataxin is not completely understood, it has structural homologues in mammals, invertebrates, plants and yeast and is believed to be involved in iron transport in mitochondria. Gene knockout of the yeast homologue results in a shift of cytosolic iron into the mitochondria, resulting in a compensatory upregulation of iron transporters in the plasmalemma, which may result in an amplification of iron deposition. Increased mitochondrial iron may lead to oxidative injury or interference with oxidative phosphorylation. Clinical trials using antioxidants and enhancers of respiratory chain function have shown some cardiac protection. The gracile and cuneate nuclei typically demonstrate obvious neuronal loss, as may the vestibular nuclei. Other cranial nerve, basal pontine and olivary nuclei are usually spared, although hypertrophic degeneration of the inferior olivary nuclei is occasionally present. Cell loss and astrocytosis are usually seen in the vestibular and cochlear nuclei and in the superior olives. Purkinje cells may demonstrate dendritic abnormalities, axonal torpedoes and terminal axonal alterations that contribute to the grumose degeneration seen around neurons in the dentate nuclei. There may be neuronal loss in the ventral-posterior thalamus, external globus pallidus and subthalamic nuclei. The cerebral cortex is spared, although there may be loss of Betz cells in the primary motor cortex. The cardiomyopathy is characterized by hypertrophy of myocytes and pleomorphic nuclei. The heart is often enlarged and has multiple foci of myofibre loss, with fibrosis and variable amounts of chronic inflammation. Pathological studies are not documented, but neuroimaging has shown atrophy, primarily in the cerebellum. Grossly, the spinal cord appears to be abnormally small, often with obvious loss of myelinated fibres in the dorsal and lateral columns. The cerebellum rarely shows gross atrophy, but the dentate nuclei may appear reduced in volume. Most cases 804 (a) Chapter 13 Degenerative Ataxic Disorders (d) (b) (e) (c) and a complex movement disorder usually dominated by chorea or dystonia. The characteristic conjunctival and cutaneous telangiectasias usually develop within a few years of the onset of ataxia. Abnormalities of the immune system, chiefly athymia or thymic hypoplasia and immunoglobulin deficiency, result in recurrent respiratory tract infections. Nearly half of patients develop a neoplasm, most commonly leukaemia or lymphoma, sometimes preceding the onset of ataxia. With increasing age, there is an increased risk of various carcinomas, malignant melanoma and primary brain neoplasms. There is an increased sensitivity to detrimental side effects of radiation exposure. Laboratory investigations show elevated alpha-fetoprotein levels, cytogenetic evidence of chromosome fragility and increased radiosensitivity in cultured lymphocytes or fibroblasts. There is myelin pallor due to axonal loss in the posterior roots and columns and pyramidal tracts. There is atrophic pallor of the posterior roots and columns, pyramidal tracts and spinocerebellar tracts. Atrophic pallor is present in the posterior roots and columns (gracile > cuneate), anterior and lateral pyramidal tracts and both posterior and anterior spinocerebellar tracts. Neuropathological studies have shown changes, such as posterior column degeneration with axonal spheroids, which can be seen in vitamin E deficiency in other clinical contexts. There is increased lipofuscin accumulation in a number of neuronal populations, including the dorsal root ganglia. As this form of ataxia is treatable with dietary vitamin E supplementation, diagnostic studies for early-onset ataxias should include vitamin E levels. The gene is large, with 150 kb and 66 exons; many different mutations have been identified throughout the gene, without clustering in mutational hot spots, so molecular diagnosis is difficult. Most of the known mutations result in either truncation of the protein or splicing abnormalities.
Procyclic trypomastigotes (bloodstream trypomastigotes multiply by binary ssion are ingested) = Infective stage = Diagnostic stage 4 Trypomastigotes in blood 21 cholesterol ratio range order 10 mg ezetimibe with amex. The parasite invades the bloodstream and infects the lymphoreticular system cholesterol hdl ratio low buy ezetimibe on line amex, with chronic lymphadenopathy and hepatosplenomegaly cholesterol medication heart attack generic ezetimibe 10 mg. The infection passes into the brain and spinal cord across the bloodbrain barrier after a few weeks exogenous cholesterol definition order 10 mg ezetimibe, and there is infection of the choroid plexus cholesterol levels reading test results buy cheap ezetimibe 10 mg on-line. Extrapyramidal signs with chorea, ataxia and muscle fasciculation lead to coma and death. Microglial activation is universal and nodules are variable; there is always a diffuse astrocytosis. Crucially, no parasites are evident on light microscopy in the neuropil, and rarely in the meningeal spaces (except in acute T. The lymph nodes show reactive follicular hyperplasia and medullary plasmacytosis, and with T. In this infection also, the heart may be involved with a chronic pancarditis and, in severe cases, a true myocarditis,147 which results in fibrosis in survivors. The parasites cross the bloodbrain barrier and induce immunopathology, which has been more studied experimentally. Also, by entering the perivascular spaces of the brain, the parasite escapes the host immune system and lysis. Note the extensive abnormal signal in the basal ganglia and periventricular zones. Protozoal Infections (a) 1249 present in blood and node, but parasites more difficult to find in patients with T. Distinction of the haemolymphatic from the cerebral stage is critical, because the toxic drugs necessary for the invasive disease are not needed for the earlier stage. Pentamidine is used for the haemolymphatic stage of infection, but is ineffective in the meningoencephalitic stage because it does not cross the bloodbrain barrier. There is also genetic polymorphism of the parasites which, coupled with evident variation in host inflammatory response genes, makes our understanding of this classical tropical disease only rudimentary at present. Inside cells they transform into amastigotes 8 Metacyclic trypomastigotes in hindgut 7 Multiply in midgut 6 Epimastigotes in midgut 5 Triatomine bug takes a blood meal (trypomastigotes ingested) = Infective stage = Diagnostic stage Trypomastigotes can infect other cells and transform into intracellular amastigotes in new infection sites. Clinical manifestions can result from this infective cycle 3 Amastigotes multiply by binary ssion in cells of infected tissues 4 Intracellular amastigotes transform into trypomastigotes, then burst out of the cell and enter the bloodstream 21. Bugs inhabit the roofs of mud houses, carry the parasites in the posterior intestine, in the form of trypomastigotes which are eliminated in the faeces when they bite, and enter at the site of the bite because of itching and scratching. This parasite evades host immune surveillance by coating its surface with microvesicles released from blood cells early during infection. Clinical Features the disease has a variable clinical course, ranging from asymptomatic infection to severe chronic disease with cardiovascular or gastrointestinal involvement or, occasionally, overwhelming acute episodes. It is directly related to the parasitaemia, may be asymptomatic or course with fever, swollen eyelids, hepatosplenomegaly and enlarged lymph nodes. About 10 per cent of the patients, especially children, develop a severe acute illness with encephalitis and myocarditis, death ensuing in virtually all cases. After recovery, patients enter an intermediate stage, when circulating parasites disappear. However, scanty parasitized cells persist as a latent infection, remaining as inflammatory foci in cardiac and extracardiac tissues. During this phase, the host immune response is assumed to suppress both recrudescence of infection and the autoimmune reactions that may characterize the chronic disease. This is mainly extracranial disease affecting gut and heart, but with some brain syndromes, including stroke, encephalopathy and reactivated acute lesions. In addition to these, tumour necrosis factor, interleukin and nitric oxide have crucial roles in host resistance to infection with T. Perivascular infiltrates of lymphocytes, some polymorphs, multiple foci of microglial proliferation, and microglial nodules are scattered within the brain parenchyma. Various neurological syndromes have been reported, including spastic paralysis, dementia, involuntary movements and cerebellar ataxia. These are relatively rare and some authors do not feel there is a distinct, separate chronic chagasic encephalopathy in non-immunocompromised patients. Astrocytes are the main target of the parasites, and neurons are spared in spite of their proximity to both parasites and nodular reaction containing macrophages. The conduction systems of the heart and autonomic plexi in the wall of the oesophagus and gut are particularly affected leading to dysfunction in peristalsis, which culminates in the development of mega-oesophagus and/or megacolon. The pathogenetic mechanisms responsible for the lesion in the cardiac autonomic nervous system are multiple, but all contribute to the final result: denervation of the organ and subsequent cardiomegaly, cardiomyopathy, arrhythmias and death. No clinical evidence of spinal motor neuron damage is found, although their involvement was suggested by the presence of some polyphasic enlarged amplitude motor unit potentials. It is likely that the number of motor neurons involved does not reach the levels needed to give overt clinical manifestations. Protozoal Infections 1253 antibodies that cross-react with host epitopes, such as neurons and myelinated nerve sheaths, a process of molecular mimicry. Encephalitis due to Sappinia diploidea Acanthamoeba, Balamuthia and Naegleria spp. This view is supported particularly because of the apparent discrepancy between the intensity of inflammatory reaction and scarce number of parasites in chronic chagasic myocarditis. Clinically, its onset is abrupt with a rapid progression to death within 72 hours if not treated. It starts as a focal neurological lesion with local meningitis, and may mimic bacterial meningitis. Most cases are diagnosed at autopsy, where it appears as a necrotic mass in the grey and white matter, up to 4 cm or larger. Histopathologically, the necrosis is featureless (but lacks polymorphs) and there are characteristic amoebic trophozoites by vessels, in the necrosis, and at the advancing edge of the lesion. The risk of infected mothers (chronic phase) transmitting the disease to their foetus during pregnancy is probably <1 per cent. The two main drugs are nifurtimox and benznidazole,131 but cure even in acute infection is only in about 70 per cent of cases. If a diagnosis can be made in time and the cerebral lesion excised, followed by treatment with metronidazole, recovery is possible. A Gram stain destroys the parasites, so a wet mount with phase contrast illumination is required; in wet preparations, the parasites appear larger at 1020 m diameter. The parasites are centred on the vessels and there is peripheral mononuclear and neutrophil inflammation. Granulomatous Amoebic Encephalitis the free living amoebae that cause this pattern of disease, less fulminant than Naegleria infection but still with high mortality, are present all around the world in soil. The differentiation is made by in vivo culture, specific immunocytochemistry and molecular diagnostic methods. The organism invades the nasal mucosa and enters the brain by travelling along the olfactory nerves. This inflammation and infection can be seen extending along the VirchowRobin spaces, associated with haemorrhagic necrosis of the brain grey and white matter, and some reactive astrocytosis if the patient has lived long enough. If the nasal mucosa is examined, there is inflammation of the epithelium extending through the cribriform plate. Clinically, the presenting features include headache, seizures, focal neurological signs and Protozoal Infections 1 Cyst 1255 21 5 4 Amoebae (cysts and trophozoites) can enter humans in various ways 6 Through nasal passages to the lower respiratory tract1 Through ulcerated or broken skin1 2 Trophozoites Cysts and trophozoites in tissue 7 = Infective stage 3 Mitosis 21. Histopathologically, the lesions are often seen to be angiocentric, with the parasites clustered around vessels that are inflamed and showing fibrinoid necrosis. The surrounding brain tissue is necrotic and shows marked astrocytosis and microgliosis. True tuberculoid granulomas are not usually seen, but characteristically there are many foreign body and Langhans type giant cells in the inflamed areas. A clue may be the presence of a chronic skin ulcer or nodule, and biopsy can provide the diagnostic clue to the brain lesion. Drug treatment, if the disease is suspected, is empirical, and many agents have been used (including amphotericin B and fluconazole), usually with little positive effect. Acanthamoeba Keratitis this is a rare sight-threatening corneal keratitis that is particularly associated with the wearing of contact lenses and with infected corneal transplants. It causes pain, conjunctivitis, photophobia, and corneal ulceration that is recurrent and relapsing. The diagnosis is made by scraping the cornea and examining for cysts and trophozoites, or by corneal biopsy. They have eosinophilic cytoplasm, pale vesicular nucleus and a prominent nucleolus. Balamuthia mandrillaris infection that spread to brain from a cutaneous traumatic lesion. Early diagnosis is important to retain sight, and treatment includes debridement of necrotic areas and application of conazole antibiotic agents. Metazoal Infections 1257 infection, the main species involved are Encephalitozoon cuniculi and Trachipleistophora anthropophthera. Microscopically, astrocytes are parasitized (but not neurons) and there is localized microglial proliferation. The parasites are seen in clusters as haematoxyphilic nuclear dots within refractive clear cytoplasm. Encephalitis due to sappinia diploidea this is a rare amoebic encephalitis that causes necrotic haemorrhagic lesions in the brain. The distinctive features are (1) the nucleus of the large (4070 m) trophozoites, which have two nuclei attached to each other by connecting filaments as seen with electron microscopy and (2) erythrophagocytosis. Cerebral microsporidiosis is always part of disseminated disease, with virtually all organs infected including gut, kidney, liver, heart and cornea. They are small 1 3 m obligate intracellular parasites that have a unique structure. The identification of species requires electron microscopy, although molecular diagnostics are increasingly used. Some affect mainly conjunctiva, producing a keratoconjunctivitis in normal as well as immunocompromised hosts. Others can cause disseminated inflammatory and necrotic lesions in all organs, including the brain, and some affect mainly skeletal muscle. For brain Metazoal InfectIons the major worm (helminth) infections that affect the nervous system of man are listed in Table 21. Traditionally, they are grouped into nematodes (roundworms) and flatworms, which comprise trematodes and cestodes. Biologically, there are three patterns of infection in man, which relate to the specificity of the human as host and how the worms mature and develop during infection. There is a wide range of other, rarer worm infections that occasionally affect man, often in single case reports only. It is impossible to differentiate between, for example, toxoplasmosis and lymphoma: autopsy proved the lesions to be B-cell lymphoma. Infection by a larval form that develops into the adult, and this produces eggs or other forms of immature worm that can be transmitted to the environment (sometimes via a vector) and so complete the infection cycle. Zoonotic (animal) disease and man gets infected accidentally, although the migration and development pattern of the parasites is comparable to that in animals. In some, the cross-sections of the polar tube can be seen under the cell membrane. These are accidentally ingested, usually by children, and hatch into larvae in the gut. The larvae have a thick pale cuticle and have induced a giant cell granulomatous reaction in the neuropil. This is not common, with only 20 neurotoxocariasis cases published between 1956 and 2004. In one highly endemic zone in Brazil, where 30 per cent of children admitted to hospital have antibodies to T. Antihelminthic treatment is with albendazole, usually plus steroid anti-inflammatory cover. Ocular Toxocariasis Children present with reduced vision, strabismus, uveitis or leukocoria. Granulomas and fibrosis may be seen, but larvae are not always identified in the specimens. Clinical Features Clinically, the initial infection may be accompanied by fever, cough and malaise; abdominal pain, hepatomegaly and pneumonitis with wheezing also occur. Other Neurological and Ocular Larva Migrans Syndromes the differential diagnosis of toxocariasis includes strongyloidiasis, angiostrongyliais, and other, rarer, visceral larva migrans syndromes, such as caused by Ascaris lum bricoides, other Toxocara species, Halicephalobus165 and Baylisacaris98 infections41 (see Other Rare Nematode Infections, later in chapter). Strongyloidiasis Strongyloides stercoralis is an intestinal infection with man as the definitive host. It distributed widely in the tropics and subtropics, but not in northwestern Europe. Adult female worms in the small bowel mucosa lay eggs that rapidly transform into larvae. These are excreted and develop further in the soil, so that infection to another person is via penetration of bare skin. Strongyloides in virtually unique in that it also has an autoinfection cycle: larvae in the bowel reinvade the host, perpetuating the infection for decades after initial exposure. Immunosuppression permits the autoinfection cycle to accelerate and produce rapidly overwhelming infection (hyperinfection syndrome) that disseminates to all organs. Pathologically, the purulent meningitis may also manifest with filariform larvae of S.
References
- Frerman FE, Goodman SI. Fluorometric assay of acyl CoA dehydrogenases in normal and mutant fibroblasts. Biochem Med 1985;33:38.
- Chang DF, Campbell JR: Intraoperative floppy iris syndrome associated with tamsulosin, J Cataract Refract Surg 31(4):664n673, 2005.
- Abdulla FA, Moran TD, Balasubramanyan S, Smith PA. Efects and consequences of nerve injury on the electrical properties of sensory neurons. Can J Physiol Pharmacol 2003;81:663-682.
- Hung SY, Yang WC, Luo HL, et al: Segmental ureterectomy does not compromise the oncologic outcome compared with nephroureterectomy for pure ureter cancer, Int Urol Nephrol 46(5):921n926, 2014.
- Zaghloul KA, Schramm J. Surgical management of glioneuronal tumors with drug-resistant epilepsy. Acta Neurochir 2011; 153(8):1551-1559.
- Mendenhall WM, Million RR, Cassisi NJ. Elective neck irradiation in squamous cell carcinoma of the head and neck. Head Neck 1980;3:15-20.
- Goulet RJ Jr, Hardacre JM, Einhorn LH, et al. Hepatic resection for disseminated germ cell carcinoma. Ann Surg. 1990;212:290-293; discussion 3-4.