

Female Cialis
| Contato
Página Inicial
Chirag M. Sandesara, MD
- Fellow, Division of Cardiology
- Department of Internal Medicine
- Roy J. and Lucille A. Carver College of Medicine
- University of Iowa
- Iowa City, Iowa
The diagnosis is easily missed since these symptoms are common in normal pregnancy menopause period changes female cialis 10 mg purchase otc. Moreover women's health center hilo purchase 20 mg female cialis visa, poorly controlled thyrotoxicosis can result in fetal tachycardia menstruation calendar discount female cialis online mastercard, intrauterine growth retardation menstruation 1800s buy female cialis 10 mg cheap, prematurity 1st menstrual period after pregnancy buy 20 mg female cialis free shipping, stillbirth and possibly even congenital malformations. Antithyroid drugs are the treatment of first choice for thyrotoxicosis in pregnancy. Newly diagnosed hyperthyroidism during pregnancy can be treated with -adrenoceptor antagonists (-blockers) in the short term, followed by antithyroid drugs. Thyroid surgery is sometimes necessary because of poor drug adherence, drug hypersensitivity or failure of medical treatment and is most safely performed during the second trimester. Radioactive iodine is absolutely contraindicated throughout pregnancy, as it invariably induces fetal hypothyroidism. Frequent review of mother and fetus (monitoring heart rate and growth) is important during pregnancy and in the puerperium. It can present with failure to establish lactation after birth, amenorrhoea or other features of hypopituitarism. The diagnosis can be confirmed by tests of pituitary function and treated with hormone replacement, as described on page 682. Women with mild disease can be managed conservatively but if serum calcium levels rise above 2. If parathyroidectomy is required, it should ideally be performed during the second trimester. Rheumatoid arthritis often improves during pregnancy, particularly in those who are negative for rheumatoid factor or anti-cyclic citrullinated peptide antibodies. There is an increased risk of pre-eclampsia, pre-term birth and small babies for women with active disease, emphasising the importance of maintaining disease control during pregnancy. Disease flares are common in the post-partum period, regardless of serology, and this can pose a problem for breastfeeding and care of the infant. Because of this, pregnant women with diabetes insipidus may need higher doses of desmopressin until delivery. Although fertility is reduced in patients with prolactinoma, pregnancies can occur and if this happens the tumour may enlarge as part of the physiological pituitary enlargement that takes place during normal pregnancy. Macroprolactinomas (10 mm) are at greater risk of enlarging and may cause optic chiasm compression. Measurement of serum prolactin is generally not helpful, since levels increase anyway as part of normal pregnancy. Dopamine receptor agonists such as cabergoline and bromocriptine should normally be stopped during pregnancy, but can be reintroduced if necessary in patients with an enlarging prolactinoma that is threatening the visual fields. Human immunodeficiency virus infection fre co m Women with known adrenal insufficiency can continue their glucocorticoid and mineralocorticoid replacement during pregnancy as normal. If this occurs, the diagnosis is challenging because total cortisol normally increases during pregnancy, and short Synacthen tests (p. There is an increased risk of pre-eclampsia, thrombosis, fetal growth restriction, pre-term delivery, miscarriage and fetal death. Although these are normally contraindicated in pregnancy, the potential benefit in this situation outweighs the risk to the fetus. Medications should be reviewed prior to pregnancy, to ensure they are safe, and an alternative substituted if necessary (see Box 30. Women who have a history of surgically corrected congenital heart disease generally tolerate pregnancy well, but are more likely to have babies with congenital heart disease and should be offered fetal cardiac scans. Acyanotic heart diseases, such as atrial septal defect, ventricular septal defect and patent ductus arteriosus, all have a good prognosis in pregnancy. Unrepaired cyanotic heart disease has a very poor prognosis in pregnancy, as does pulmonary hypertension, regardless of the underlying cause. If necessary, warfarin can be used during pregnancy, particularly in the second and third trimesters. It is a diagnosis of exclusion, made when other causes of heart failure have been ruled out. There is a significant chance of reduction in cardiac function in subsequent pregnancies. Women with regurgitant lesions, such as mitral regurgitation and aortic regurgitation, tolerate pregnancy better than those with stenotic lesions. Mitral stenosis causes a reduction in blood flow from the left atrium to left ventricle in diastole, which worsens during pregnancy due to the increased heart rate and hypervolaemia. Surgical intervention is indicated if there is continued haemodynamic compromise despite optimal medical management. If asymptomatic bacteriuria is discovered during pregnancy, it should be treated promptly with antibiotics, to prevent ascending renal tract infection. Pyelonephritis is more common in pregnancy due to the physiological dilatation of the upper renal tract; if it does occur, it can trigger premature labour. While atherosclerosis is the main cause in non-pregnant individuals, coronary artery dissection and coronary thrombosis secondary to the hypercoagulable state are more common causes during pregnancy. Clopidogrel can be given but should be stopped around the time of delivery to reduce the risk of uterine bleeding and to allow spinal anaesthesia to be used if necessary. Stenting can be performed, but bare-metal stents are preferred because drug-eluting stents require dual antiplatelet therapy that cannot be continued around the time of delivery. Proteinuria caused by glomerular disease is usually exacerbated during pregnancy, and nephrotic syndrome may develop without any alteration in the underlying disease activity in individuals who had only slight proteinuria before pregnancy. This further increases the risk of venous thromboembolism, the leading cause of maternal deaths in developed countries. The outcome is best for women with a well-functioning graft, with no proteinuria or hypertension. Women with renal transplants can deliver vaginally but in practice there is a higher incidence of caesarean section in this group, due to the higher incidence of pre-term delivery. It is more common in first pregnancies and multiple pregnancies, and is associated with male fetuses. A liver biopsy is rarely needed to make the diagnosis but shows microvascular steatosis. More intensive dialysis is recommended in pregnancy, and particular attention should be paid to addressing issues around blood pressure, fluid balance and anaemia. Acute fatty liver of pregnancy can be diagnosed when 6 of the above features are present in the absence of another explanation. It usually presents antenatally but can also appear for the first time in the postnatal period. The presenting symptoms can be the same as those of pre-eclampsia but can also include headache, right upper quadrant pain and visual disturbance. Management involves supportive care, control of hypertension, correction of coagulopathy and delivery of the fetus. In most cases, it responds well to oral iron supplementation, with a rise in haemoglobin of approximately 0. If the haemoglobin does not rise following a 4-week trial of iron supplementation, alternative causes of anaemia should be considered. Non-adherence to oral iron is common and intravenous iron should be considered in women with iron deficiency and failure of oral treatment. It is generally not necessary to investigate iron deficiency anaemia during pregnancy unless there is clinical evidence of gastrointestinal blood loss, which should be investigated in the normal way. Vaccinations and immunoglobulin should be given to infants of mothers who test positive for hepatitis B, and antiviral agents should be given to the mother after delivery. Pregnant women are at greater risk of contracting hepatitis E than the non-pregnant population. It is transferred via the faeco-oral route, and is usually a mild self-limiting illness outside of pregnancy. Haematological disease co Mood changes are common during pregnancy but more severe psychiatric disorders, such as depression or psychosis, typically present within 24 weeks of delivery. The cause is incompletely understood but the condition is thought to be due in part to the cholestatic effect of high oestrogen levels. The typical presentation is in the third trimester with pruritus, particularly affecting the soles and palms. The diagnosis can be made on the basis of these clinical features when other causes of liver dysfunction and pruritus have been excluded. Treatment is with ursodeoxycholic acid in a starting dose of 250 mg twice daily, which usually improves symptoms and liver function. There is an increased risk of fetal mortality with evidence of a particularly high risk when bile acid levels are over 40 µmol/L (97. Treatment therefore aims to bring bile acids below 40 µmol/L and some centres induce labour before 40 weeks in an effort to reduce the risk. Treatment with acetazolamide can be continued during pregnancy but should be avoided in the first trimester due to lack of safety data. While pregnancy does not generally affect the frequency of seizures in women with well-controlled epilepsy, those who enter pregnancy with poorly controlled epilepsy are likely to deteriorate. Seizures are more common at the time of delivery and women should be advised to deliver in a unit staffed with personnel able to manage this. The management of stroke during pregnancy is similar to that in non-pregnant patients. The presentation is with headache, seizures and neurological deficits such as hemiparesis. Management of acute infarct should be as for the non-pregnant patient and include consideration of thrombolysis. If necessary, prophylaxis can be given with aspirin, -blockers or tricyclic antidepressants. Triptans can therefore be used for the treatment of migraine if other therapies are ineffective. The most common cause is gestational thrombocytopenia, which typically occurs towards the end of pregnancy and resolves spontaneously after delivery. It is not associated with adverse pregnancy outcomes and requires no specific intervention. This should be managed with glucocorticoids and/or immunoglobulin, with the aim of maintaining the platelet count above 80 × 109/L at the time of delivery, in case spinal anaesthesia or caesarean section is required. These conditions are rare but important to recognise since up to one-quarter of cases occur during pregnancy and the post-partum period. A useful evidence-based guideline on the investigation of pulmonary embolism in pregnancy. Measurement of D-dimer is not useful in pregnancy because levels rise as part of normal pregnancy. Improve outcome co Parents oversee care Loss of parental control Patient takes responsibility m Survivors of previously lethal conditions Ongoing medical problems, or complications of previous therapy Live with family Leave home Independent living m ok Transition planning Pre-pubertal Puberty ok s ok ok eb New clinical specialties. Education Graduation co Employment Financially independent m Dependent Transition Autonomous m eb · Osteogenesis imperfecta · Hypophosphataemic rickets Paediatric services are organised and delivered in a very different way to adult medical services. Most research in this field has been undertaken with young people with diabetes, and many of the outcome measures relate to that condition. Adult medicine traditionally comprised patients with progressive conditions, and increasing pathology with advancing age. Common illnesses include asthma, epilepsy, congenital heart disease, diabetes and childhood cancer (Box 31. Planning the process of transition from paediatric to adult health services and improving the assessment of young people as they enter those adult services have been shown to impact positively on long-term health outcomes. There is a need for physicians to gain new skills in the care of young people and adults who have conditions that have arisen in childhood. This includes developing specific skills in the management of adolescents and young adults, managing the process of transition and developing knowledge of relevant medical conditions. The overall approach to transition medicine, as well as important disease-specific issues, will be considered in this chapter. Once puberty has been completed, teenagers can be considered, in pharmacokinetic and pharmacodynamic terms, to behave like adults. At the same time as undergoing transition within medical services, young people are making multiple other transitions in their lives as they move from a dependent to an independent way of living. They often move away from the family home, and parents who formerly held responsibility for patient management, coordination of care, communication and consent to treatments will be demoted to an advisory role. Paediatric services are not well placed to meet this change in focus from the patient as a child to the patient as an independent adult, and young people benefit from the move to adult services as long as their specific needs as a young adult are recognised. The first step is to establish a policy in consultation with young people and train staff in the policy. Subsequently, systems need to be developed to identify patients in need of transition and track them as they pass through the programme. Adult health-care providers need to be identified and processes developed for introducing the young person to the adult team. Communicate with paediatrics and confirm transfer Help young adult to access other adult services Continue individualised care plan tailored to young person Seek feedback from young adult about transition co co m Integration into adult services ok s ok ok ok eb ks 1. Current and future education/impact of condition on career plans School attendance and performance Work experience and access to careers advice Outside activities and interests Disclosure to school/employer. Self-care/meal preparation Independent travel/mobility Trips/overnight stays away from home Benefits/financial independence m (A) Activities of daily living eb · Establish process to identify patients · Develop systems to track individual progress · Incorporate transition planning into clinical care 1. Understands sexual health issues/pregnancy/sexually transmitted infections oo k oo oo ks Tracking and monitoring (H) Health and lifestyle sf re e Establishing transition policy. Can attend part/whole clinic appointment on their own Knows how to make appointments/alter appointments Has understanding of confidentiality Orders repeat prescriptions Takes some/complete responsibility for medication/other treatment Knows where to get help om m eb oo 31. The general advice is that when prescribing a dose per kilogram, the optimal weight for height rather than actual weight should be used for obese young people.
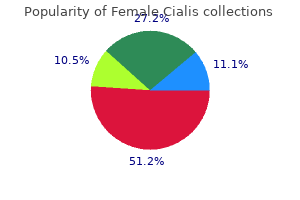
Human albumin 20% solution is used in the management of hypoproteinaemic oedema in nephrotic syndrome (p women's health clinic jeffersonville indiana 20 mg female cialis purchase overnight delivery. Stroke breast cancer 2014 game cheap 10 mg female cialis mastercard, transient ischaemic attacks menopause questionnaire female cialis 20 mg order on-line, amaurosis fugax menstruation rituals around the world discount female cialis 20 mg online, digital ischaemia or gangrene breast cancer oncologist purchase female cialis online pills, aquagenic pruritus, splenomegaly and systemic upset are also features. Patients with myeloproliferative disorders may also present with features such as aquagenic pruritus, splenomegaly and systemic upset. Relatively minor symptoms of transfusion reactions (fever, itch or urticaria) occur in up to 3% of transfusions, and usually in patients who have had repeated transfusions. Any symptoms or signs that arise during a transfusion must be taken seriously, as they may be the first warnings of a serious reaction. These chains are attached to proteins and lipids that lie in the red cell membrane. Pooled units (of 10 donations) will raise fibrinogen by 1 g/L von Willebrand disease and haemophilia If virus-inactivated or recombinant products are not available. Following delivery of an RhD-positive baby, the mother is given further anti-D within 72 hours; a maternal sample is checked for remaining fetal red cells and additional anti-D is given if indicated. All these substances may lead to inflammation, increased vascular permeability and hypotension, which may, in turn, cause shock and renal failure. Inflammatory mediators can also cause platelet aggregation, lung peribronchial oedema and smooth muscle contraction. RhD-negative individuals do not normally produce substantial amounts of anti-RhD antibodies. However, if RhD-positive red cells enter the circulation of an RhD-negative individual, IgG antibodies are produced. This can occur during pregnancy if the mother is exposed to fetal cells via fetomaternal haemorrhage, or following transfusion. If a woman is so sensitised, during a subsequent pregnancy anti-RhD antibodies can cross the placenta; if the fetus is RhD-positive, haemolysis with severe fetal anaemia and hyperbilirubinaemia can result. Therefore, an RhD-negative female who may subsequently become pregnant should never be transfused with RhD-positive blood. In RhD-negative women, administration of anti-RhD immunoglobulin (anti-D) perinatally can block the immune response to RhD antigen on fetal cells and is the only effective product for preventing the development of Rhesus antibodies (Box 23. These antigens include Rhc, RhC, RhE, Rhe, and the Kell, Kidd and Duffy antigen systems. However, some patients who received transfusions before these tests were available suffered serious consequences from infection; this serves as a reminder to avoid non-essential transfusion, since it is impossible to exclude the emergence of new or currently unrecognised transfusion-transmissible infection. Prevention is by gamma- or X-ray irradiation of blood components before their administration to prevent lymphocyte proliferation. Any antibody detected can be identified by further testing, so that red cell units that lack the corresponding antigen can be selected. Full cross-matching takes about 45 minutes if no red cell antibodies are present, but may require hours if a patient has multiple antibodies. Hospitals should have local major haemorrhage protocols and all clinical staff must be familiar with their content. Good team working and communication are essential to prevent poor clinical outcome, suboptimal or inappropriate transfusion practice and component wastage. This allows group-specific units to be issued quickly and safely, for elective and emergency transfusion. Most incompatible transfusions result from failure to adhere to standard procedures for taking correctly labelled blood samples from the patient and ensuring that the correct pack of blood component is transfused into the intended patient. Every hospital where blood is transfused should have a written transfusion policy used by all staff who order, check or administer blood products. Platelets should be kept above 50 × 109/L; to allow for delivery time, platelets should be requested if there is ongoing bleeding and the platelet count has fallen below 100 × 109/L. Many haematological malignancies are sensitive to the effects of chemotherapy drugs and, as such, chemotherapy is the mainstay of treatment for most haematological cancers. Despite cancer cells being more sensitive, chemotherapy is largely non-specific and kills some normal cells as well as cancer cells. This leads to common side-effects of treatment, such as transient bone marrow failure, mucositis and infertility. The supportive care of patients undergoing chemotherapy is critical in overcoming these side-effects. It is this supportive care, including blood product support, antibiotics, antifungal drugs, growth factors and antiemetics, that has allowed specialist haematology units to achieve the best possible results from intensive chemotherapy: for example, when treating acute leukaemia. The basic principles of chemotherapy include combining several non-cross-reacting drugs in a regimen that kills a fixed proportion of cancer cells with a given dose. Several cycles of the combination are given to achieve gradual reduction of the tumour burden, to induce remission and, in some instances, to produce a cure (p. Chemotherapy drugs can also be linked to a monoclonal antibody to allow fre eb oo ks ks fre. Small molecules targeted at the mechanisms causing cancer are replacing chemotherapy in some disease situations, such as tyrosine kinase inhibitors in chronic myeloid leukaemia and inhibitors of B-cell signalling in relapsed chronic lymphocytic leukaemia and lymphomas. During this period of aplasia, patients are at risk of infection and bleeding, and require intensive supportive care as described on page 957. It may take several years to regain normal immunological function and patients remain at risk from opportunistic infections, particularly in the first year. Reduced-intensity conditioning has enabled treatment of older or less fit patients. Such transplants have produced long-term remissions in some patients with acute leukaemia and myelodysplastic syndromes aged 4065 years, who would not previously have been considered for a myeloablative allograft. The risks and outcomes of transplantation depend upon several patient- and disease-related factors. After conditioning myeloablative therapy, the autologous stem cells are reinfused into the blood stream in order to rescue the patient from the marrow damage and aplasia caused by chemotherapy. It can affect the skin, causing rashes, the liver, causing jaundice, and the gut, causing diarrhoea, and may vary from mild to lethal. Severe presentations are very difficult to control and, despite high-dose glucocorticoids, may result in death. It often resembles a connective tissue disorder, although in mild cases a rash may be the only manifestation. The guiding principles are outlined here but management in specific indications is discussed elsewhere in the book. Thus, antiplatelet agents, such as aspirin, clopidogrel and, increasingly, ticagrelor, are the drugs of choice in acute coronary events (p. In some extremely prothrombotic situations, such as coronary artery stenting, a combination of anticoagulant and antiplatelet drugs is used (p. A wide range of anticoagulant and antithrombotic drugs is used in clinical practice. Newer agents allow predictable anticoagulation without the need for frequent monitoring and dose titration. This results in platelet activation and a prothrombotic state, with a paradoxical thrombocytopenia. Warfarin anticoagulation typically takes more than 35 days to become established, even using loading doses. Patients who require rapid initiation of therapy may receive higher initiation doses of warfarin. The major problems with warfarin are: · a narrow therapeutic window · metabolism that is affected by many factors · numerous drug interactions. This assigns a score based on: · the thrombocytopenia · the timing of the fall in platelet count · the presence of new thrombosis · the likelihood of another cause for the thrombocytopenia. The patient may be asymptomatic, or develop venous or arterial thrombosis and skin lesions, including overt skin necrosis. Affected patients may complain of pain or itch at injection sites and of systemic symptoms, such as shivering, following heparin injections. Patients who have received heparin in the preceding 100 days and who have preformed antibodies may develop acute systemic symptoms and an abrupt fall in platelet count in the first 24 hours after re-exposure. The key features of these drugs include the fact that they are efficacious in fixed oral doses, have a short half-life of around 10 hours, achieve peak plasma levels Blood loss the most common explanation in men and post-menopausal women is gastrointestinal blood loss (p. This may result from occult gastric or colorectal malignancy, gastritis, peptic ulceration, inflammatory bowel disease, diverticulitis, polyps and angiodysplastic lesions. Worldwide, hookworm and schistosomiasis are the most common causes of gut blood loss (pp. Major bleeding is the most common serious side-effect of warfarin and occurs in 12% of patients each year. There are scoring systems that predict the annual bleeding risk and these can be used to help compare the risks and benefits of warfarin for an individual patient (Box 23. If the patient is not bleeding, it may be appropriate to give a small dose of vitamin K either orally or intravenously (12. Red cells in the bone marrow must acquire a minimum level of haemoglobin before being released into the blood stream. While in the marrow compartment, red cell precursors undergo cell division, driven by erythropoietin. A similar defect of cell division is seen in the presence of cytotoxic drugs or haematological disease in the marrow, such as myelodysplasia. The red cell membrane is composed of a lipid bilayer that will freely exchange with the plasma pool of lipid. Reticulocytes are larger than mature red cells, so when the reticulocyte count is raised . An initial perceived drawback was the lack of specific reversal agents for these drugs but idarucizumab is a monoclonal antibody now available for the reversal of dabigatran, and andexanet alfa, a site-inactivated Xa molecule, is close to licensing for the reversal of apixaban and rivaroxaban (see Box 23. The general perception at present is that in these indications they are at least as efficacious as dose-adjusted coumarin and probably associated with less clinically significant bleeding. In women of child-bearing age, menstrual blood loss, pregnancy and breastfeeding contribute to iron deficiency by depleting iron stores; in developed countries, one-third of pre-menopausal women have low iron stores but only 3% display iron-deficient haematopoiesis. Gastric acid is required to release iron from food and helps to keep iron in the soluble ferrous state. Achlorhydria in the elderly or that due to drugs such as proton pump inhibitors may contribute to the lack of iron availability from the diet, as may previous gastric surgery. Iron is absorbed actively in the upper small intestine and hence can be affected by coeliac disease (p. In pregnancy, iron is diverted to the fetus, the placenta and the increased maternal red cell mass, and is lost with bleeding at parturition (Box 23. It is a very specific test; a subnormal level is due to iron deficiency or, very rarely, hypothyroidism or vitamin C deficiency. Ferritin levels can be raised in liver disease and in the acute phase response; in these conditions, a ferritin level of up to 100 µg/L may still be compatible with low bone marrow iron stores. Ferroportin available co m Inflammatory cytokines induce hepcidin secretion from liver Anaemia Hypoxia Low iron stores suppress hepcidin secretion from liver m m. The transport of iron is regulated in a similar fashion to eb oo ks ks oo k oo eb eb 23. Plasma iron has a marked diurnal and day-to-day variation and becomes very low during an acute phase response but is raised in liver disease and haemolysis. Levels of transferrin, the binding protein for iron, are lowered by malnutrition, liver disease, the acute phase response and nephrotic syndrome, but raised by pregnancy and the oral contraceptive pill. All proliferating cells express membrane transferrin receptors to acquire iron; a small amount of this receptor is shed into blood, where it can be detected in a free soluble form. At times of poor iron stores, cells up-regulate transferrin receptor expression and the levels of soluble plasma transferrin receptor increase. This oo oo eb o eb eb Investigation of the cause this will depend on the age and sex of the patient, as well as the history and clinical findings. In men and in post-menopausal women with a normal diet, the upper and lower gastrointestinal tract should be investigated by endoscopy or radiological studies. Serum anti-transglutaminase antibodies and possibly a duodenal biopsy are indicated (p. Current guidelines suggest exclusion of coeliac disease by antibody testing at an early stage of investigation. In difficult cases, it may still be necessary to examine a bone marrow aspirate for iron stores. Examination of the marrow may ultimately be required to assess iron stores directly. Trials of higher-dose intravenous iron are under way to try to bypass the hepcidin-induced blockade. Hepcidin binds to ferroportin on the membrane of iron-exporting cells, such as small intestinal enterocytes and macrophages, internalising the ferroportin and thereby inhibiting the export of iron from these cells into the blood. The iron remains trapped inside the cells in the form of ferritin, levels of which are therefore normal or high in the face of significant anaemia. Inhibition or blockade of hepcidin is a potential target for treatment of this form of anaemia. The serum iron is low but iron stores are normal or increased, as indicated by the ferritin or stainable marrow iron. Ferrous sulphate 200 mg 3 times daily (195 mg of elemental iron per day) is adequate and should be continued for 36 months to replete iron stores. Many patients suffer gastrointestinal side-effects with ferrous sulphate, including dyspepsia and altered bowel habit. When this occurs, reduction in dose to 200 mg twice daily or a switch to ferrous gluconate 300 mg twice daily (70 mg of elemental iron per day) or another alternative oral preparation should be tried.
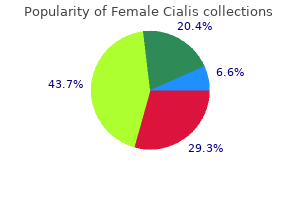
Again breast cancer gene discount female cialis 10 mg with amex, if parental chromosome studies cannot be performed first breast cancer in teens purchase female cialis 10 mg mastercard, couples with such a history should be offered prenatal diagnosis women's health center colorado order 10 mg female cialis amex. Abnormal parental karyotype (other than a balanced structural rearrangement) If one of the parents has a trisomic mosaicism pregnancy x ray lead apron buy female cialis toronto, or a sex-chromosome aneuploidy that does not abrogate reproductive ability best women's health tips female cialis 20 mg order. Trisomy 21 mosaicism has been found in both mothers and fathers of patients with Down syndrome. Prenatal cytogenetic diagnosis should therefore be offered when an individual has been established as being mosaic. Prenatal sex determination for X-linked disorders When a mother is an obligate or a proven carrier of an X-linked disease, or is at high risk for being a carrier, prenatal sex determination is justified. Fetal sexing becomes obsolete for X-linked disorders when a definitive prenatal diagnosis becomes available. However, prenatal sex determination might be sufficient when the cost of a direct test for the disorder is very high or when information about the carrier status of female offspring is not sought. One study reported a 20 percent maternal cell contamination rate in noncultivated amniotic fluid cells Reduced ovarian complement Surgical removal of part, or all, of an ovary or congenital absence of an ovary is associated with early menopause. Miscellaneous It remains to be determined whether maternal exposure to radiation before conception increases the frequency of aneuploid progeny. Several investigations have suggested some association, but none has shown statistical significance. Histories of cancer chemotherapy and other environmental exposures to mutagens are often a source of concern to patients. These patients should be counseled carefully about the limitations of prenatal cytogenetic diagnosis in order to avoid creating a false sense of security. Finally, anxiety is often used as an indication for prenatal diagnosis when there is no other medical indication for testing. Noninvasive prenatal testing should be helpful in alleviating much of this anxiety. As with concerns about mutagens, couples with this request must be fully informed about the limitations of prenatal diagnosis. Interpretation issues: chromosome mosaicism and pseudomosaicism General considerations Mosaicism involving gain or loss of a chromosome can arise by one of two mechanisms. The second mechanism involves a euploid postzygotic (somatic) cell experiencing a malsegregation event resulting in an extra copy of a chromosome in one daughter cell and a monosomy in the other. Uniparental disomy can again arise if there is a reduplication of the chromosome in the monosomic cell or loss of the appropriate chromosome in the trisomic line. Monosomy involving an autosome is generally associated with cell nonviability; therefore, only two cell lines are usually observed: the original euploid population plus the trisomic line. Within the placenta and within the fetus, further nonrandom distributions of normal and abnormal cells may occur. There may also be cell-selection pressures that favor the proliferation of normal diploid cells, relative to the proliferation of the trisomic line (or vice versa). It is known from studies on preimplantation embryos that an extremely high percentage show mosaicism and therefore strong selective pressures must exist during early development. The phenotype associated with any particular type of mosaicism can be expected to be highly variable, reflecting the differences in the proportions of normal and abnormal cells. In general, the fetal abnormalities that might be present in mosaic cases can be expected to be consistent with that seen in nonmosaic trisomy or partial trisomy. However, long-term cultures generally will be reflective of the mesenchymal core of the villi. Analysis of metaphase trophoblastic cells has the advantage of providing a quick result with minimal issues with maternal cell contamination. On the other hand, because villi mesoderm represent a cell population that is developmentally closer to the fetus itself,291 and because the quality of chromosome preparations is generally higher, the analysis of longer term culture is the preferred approach. The presence of two or more metaphase cells with the same karyotype is considered evidence for a secondary cell line provided they are found in more than one cell preparation. If, after the expanded analysis, the abnormality is confined to a single cell ("single cell pseudomosaicism"), the finding is usually considered to be insignificant. In 15 of the 38 cases, an analysis of amniotic fluid, fetal blood, or fetal tissue was carried out and there were no cases of the abnormality confirmed. However, as discussed below, the need to pursue multiple cell pseudomosaicism should be based on the specific chromosome involved. The detection of mosaicism, or suspected mosaicism, should always prompt a very careful consideration about the need for confirmatory amniocentesis. Fluorescence in situ hybridization analysis on interphase cells is also sometimes used to establish the presence of the secondary cell line. A commonly used classification system expresses the various combinations of abnormal and normal cell lineages according to their presence in trophoblasts, villi mensenchyme, and fetus (or amniotic fluid). Confined placental mosaicism was found in 1 percent and was the main cause of a discordant result. It was concluded that the direct method should not be used as the sole diagnostic technique. The mosaic subgroup included 19 percent (17/91) where there was an apparent complete discordance between the trophoblast and mesenchyme cell lineages. Absence of normal cells within trophoblasts or within mesenchyme was generally associated with a higher probability of confirmation. There were also clear differences depending on the chromosome involved; potentially viable aneuploidies such as trisomy 21, trisomy 18, and sex chromosome aneuploidies have relatively high rates of being confirmed 31. This included two cases of trisomy 21 and two cases of trisomy 18 that were not confirmed as abnormal at amniocentesis. Reliance on cultures could also lead to rare false-negative results, including some that could be attributable to maternal cell contamination (unless this is explicitly excluded by supplemental testing), and also a failure to identify cases at risk for uniparental disomy. A diagnosis of true mosaicism should be made only when two cell populations with different karyotypes are found in multiple (at least two) independent culture vessels. For in situ harvesting, the finding of an identical aneuploidy in cells from one, or more, colonies from a minimum of two different culture vessels should be the major criterion for the diagnosis of true chromosome mosaicism. Two aneuploid colonies in the same culture do not establish a diagnosis of mosaicism because cell migration can occur within a culture vessel. With in situ harvesting, there can be three different types of chromosome pseudomosaicism: (i) one cell or one region in a colony with an abnormal karyotype; (ii) all the cells of an entire single colony with an identical aberrant karyotype; and (iii) multiple colonies within the same culture vessel showing an identical abnormal karyotype. For example, examination of 14 colonies or 14 fetal cells can detect only 20 percent of chromosome mosaicism at the 95 percent confidence level. However, it is applicable for the in situ method, where N refers to the number of colonies rather than to the number of cells to be examined. A table also exists for the situation in which part of the analysis is based on colonies and part on the flask method. The frequency of pseudomosaicism with multiple cells showing an identical abnormality but restricted to one culture vessel ranged from 0. The occurrence of a single cell or a single colony with an aberrant karyotype is not at all rare, ranging from 2. The table provides the level of mosaicism that is excluded with the given confidence level when N cells are counted. To determine the number of cells to count to exclude a specific level of mosaicism (x% or greater), choose the lowest value N for which x% appears in the appropriate column. For example, to exclude 10 percent mosaicism with 90 percent confidence, 22 cells must be counted; for 95 percent confidence, 29 cells are needed, and for 99 percent confidence, 44 cells. In the structural category, there were more balanced reciprocal translocations than deletions. For example, if there are six clones in the flask (k = 6); to detect 50 percent mosaicism at 95 percent confidence level, eight cells must be analyzed. This number is obtained by looking down the column corresponding to k = 6 and reaching the first percentage not greater than 50 (shown in bold). Given that true mosaicism can never be entirely excluded, there is a concern that clinically significant abnormalities might be inappropriately dismissed as pseudomosaicism. Furthermore, true mosaicism involving a structural abnormality is relatively rare (see below). In the case of balanced rearrangements, the risk associated with a true mosaicism would be minimal. It is likely that some of the other abnormalities characterized as pseudomosaicism are similarly derived from extrafetal tissues. Abnormalities that are classified as pseudomosaicism should not therefore be viewed solely as artefacts arising during cell culture. There is no direct evidence that trisomy 2 or trisomy 7 pseudomosaicism detected in amniocytes is asso- ciated with an adverse outcome. However, as discussed below in the section on true autosomal mosaicism, some pseudomosaicism trisomies do need to be considered very carefully; there is a special concern for low-level trisomy 16 mosaicism or pseudomosaicism that may well be clinically significant. Based on the phenotypes associated with nonmosaic gain of a sex chromosome, the clinical consequences of this type of true mosaicism would be minimal. Loss of a sex chromosome may well reflect random somatic cell loss and is quite common. Concerns about pseudomosaicism need to be considered in the context of the patient anxiety that the information will cause. In the absence of direct evidence that a particular pseudomosaicism is clinically significant, it is justifiable and appropriate to interpret the results as part of a range of cytogenetic diversity that is expected to be present in a normal population. Mosaicism involving gain of an autosome: data for individual chromosomes In evaluating the risk associated with the prenatally detected rare autosomal trisomies, it is necessary to rely heavily on published case reports. For most chromosomes, the cases included in this compilation did not appear to be subject to a strong ascertainment bias that is associated with referral through ultrasound detection of fetal anomalies. However, the data are subject to publication bias; only the cases with the most unusual findings or comprehensive follow up tend to be published. For the assessment of risk for the common autosomal mosaicisms (chromosomes 13, 18, and 21) the summary of risk is based on a survey in which cases ascertained because of abnormal ultrasound findings were specifically excluded. Chromosome 1 Nonmosaic trisomy 1 appears to be incompatible with even rudimentary fetal development,317 but rare cases of trisomy 1 mosaicism have been described. There are also several reports of the expression of recessive disorders due to homoallelism in individuals with upd(2). The phenotype may include minor craniofacial anomalies, digital anomalies, wide-spaced nipples, ventriculomegaly, and developmental delay. Postnatal detection of trisomy 3 mosaicism has been described (reviewed by Sheath et al. Chromosome 4 Prenatal diagnosis of trisomy 4 mosaicism seems to be extremely rare. Among the cases with abnormal outcomes, craniofacial dysmorphism, cardiac defects, and abnormalities of hands and feet may be the most common findings. Some individuals with SilverRussell syndrome have upd(7)mat and there are imprinted genes at 7p11. Chromosome 8 In contrast to the situation seen in spontaneous abortion tissues, the additional chromosome 8 present in viable mosaic and nonmosaic trisomy 8 pregnancies is usually mitotic in origin. It is known that a clinical diagnosis of trisomy 8 mosaicism syndrome is difficult because of the subtle abnormalities associated with this disorder. What appears to be the low risk of an abnormal outcome or the high probability of finding a grossly normal appearing offspring following a prenatal diagnosis of 46/47,+8 in amniocytes may be explained by the difficulty in recognizing these subtle clinical features. In cases with fibroblasts and/or placental studies, trisomy 8 mosaicism was confirmed in the majority of cases (10 of 13 cases, 77 percent). However, there are numerous literature cases of true fetal mosaicism and therefore this finding should be pursued through additional prenatal testing (amniocentesis and ultrasound). Comparison of the major phenotypic abnormalities noted in the prenatal versus the postnatal cases show rather comparable features. Of 22 cases with successful cytogenetic follow-up studies, trisomy 9 mosaicism was confirmed in the majority of cases (73 percent). Neonatal or early infant death with multiple abnormalities appears to characterize trisomy 10 mosaicism. Only four second-trimester cases are known,327, 374, 387 all of which had normal pregnancy outcomes. Of nine grossly normal liveborns, four had follow up from 2 months to 5 years; all were reported to be developmentally normal. The overall cytogenetic confirmation rate was 70 percent from fibroblasts and/or placental tissues. This survey excluded cases with prior abnormal ultrasound findings and other factors that would constitute ascertainment bias. Abnormal outcomes were noted in 10 (40 percent), with a higher probability of abnormality when the proportion of abnormal cells was high. The overall rate of positive confirmatory studies was six of 13 cases (46 percent). There were four cases in which trisomy 13 mosaicism was diagnosed at amniocentesis (mean proportion of abnormal cells 9 percent) and the pregnancy was continued. In all four cases there were no abnormalities noted at birth and, in the three cases with follow-up cytogenetic analyses, no abnormal cells were detected. Chen reviewed an additional eight published cases of trisomy 13 mosaicism and also concluded that the spectrum of phenotype variability is broad. In two abortuses, multiple congenital anomalies and facial dysmorphisms were noted. Upd(15)mat is a cause of PraderWilli syndrome and upd(15)pat is associated with Angelman syndrome. Two abortuses showed upd(15)mat, which would be consistent with PraderWilli syndrome had these pregnancies been continued. Chromosome 16 Although there is now a substantial number of documented prenatal diagnoses of trisomy 16 mosaicism, this diagnosis is associated with a highly variable set of pregnancy outcomes and counseling is therefore complex. Although it has been predicted that imprinted genes exist on chromosome 16, none has yet been identified. However, after excluding case reports that may show ascertainment bias, this association lacked statistical significance. These data would be consistent with the hypothesis that there is substantial selection against trisomic cells, reducing the size of the early embryonic cell pool.
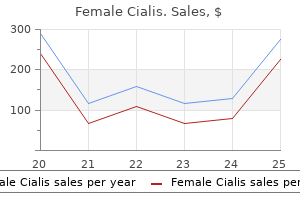
10 mg female cialis order fast delivery. Proverbs in English and Tamil - Part1.
However breast cancer 60 mile 3 day purchase discount female cialis, faced with an intractable patient women's health center perth buy female cialis 10 mg online, some guidance about disclosure is reflected in a statement issued by the American Society of Human Genetics in 1998 women's health clinic andrews afb 10 mg female cialis otc. The vast majority of medical geneticists who decided not to warn such relatives were concerned by patient confidentiality issues and legal liability pregnancy jokes humor female cialis 10 mg order on-line. Although communication and support are both vital during those fateful days breast cancer vs prostate cancer generic female cialis 10 mg with mastercard, the physician needs to recognize the great difficulty that anguished patients would have in assimilating or comprehending even the essence of any counseling. Parental counseling Physicians/counselors have a duty to convey information about the known options, risks, benefits, and foreseeable consequences133135 to couples with increased risks of having children with genetic disorders. Such a duty may be difficult, if not impossible, to fulfill if only one member of the couple attends genetic counseling. The issues are usually complex and are frequently compounded by feelings of guilt and by ignorance, family prejudices, religious obstacles, fear, and serious differences of opinion between partners. Hence, when possible (at the time the appointment is made would seem to be best), the necessity that the couple attend together should be emphasized. Not even letters written to couples after the counseling session245 (a recommended procedure, to summarize the essence of the counseling provided) can safely substitute for face-to-face discussions with both, allowing for questions and interchange about the issues and an opportunity to examine the partner. Genetic counselors should be cognizant of the complex interactive factors involved in parental reproductive decision making. Frets246 confirmed the importance of the burden of the disease in question and found that the interpretation of risk (high or low) and the wish to have children were paramount factors. The absence of personal experience of the disease was also found to be a significant influence. Frets identified a number of factors that were independently and significantly associated with problems experienced by 43 percent of counseled couples. These included no postcounseling support, recognition of high risk, disapproval by relatives, the presence of an affected child, and decisions not to have a (or another) child. Due diligence is necessary for the partners of genetic disease carriers who clearly experience significant psychologic distress. We maintain that members of the health professions should adopt as a guiding principle the critical imperative that the concept of genetic counseling be introduced in high school and in continuing public education247251 about genetic disease. Children sensitized in school about the importance of the family history, elements of heredity, concepts of individual susceptibility, and risk and opportunities for anticipatory prevention of unnecessary catastrophes, are likely to better comprehend pregnancy risks and options. Genetic counseling and prenatal diagnostic services are of little avail if many women attend for their first antenatal visit after 16 weeks of gestation. Currently, this is the case in many urban hospitals in the Western world, where between 20 and 40 percent of obstetric patients arrive at this late stage. Education beginning in high school and continued by public health authorities could effectively communicate the critical importance of preconception and prenatal care. Pelias pointed to a 1971 lawsuit257 in which the University of Chicago failed to notify women who had been given diethylstilbestrol. The university had apparently become aware of the dangers of this drug but had delayed notification for 45 years. In yet another case, after a single visit to her gynecologist for insertion of an intrauterine device (a Dalkon shield), a woman sued this physician for failing to notify her of the subsequently recognized risks of this device. Pelias253 opined that this recommendation should be recorded in clinical notes and echoed in letters to referring physicians and patients alike. However, given that tens of millions change their addresses annually and frequently seek other medical care, the patients themselves, once informed of potential advances and the need to remain in contact with a clinical geneticist, take on personal and primary responsibility. Do no harm the classic exhortation primum non nocere (first, do no harm) is as pertinent to clinical genetics as it is to medicine in all specialties. Attention to this principle arises particularly in the context of predictive genetic diagnosis, possible for a rapidly escalating number of neurodegenerative disorders. Huntington disease; some of the spinocerebellar ataxias), cardiovascular and other serious disorders including multiple endocrine neoplasia type 2B, and breast, colon and other malignancies. Published recommendations and guidelines259 urge rigorous pretest and post-test genetic counseling and recommendations that testing of children younger than 18 years of age be proscribed, except in life-threatening disorders. The inherent harm that could potentially be done by presymptomatic testing is the potential for demoralization and depression with possible suicidal consequences. Extreme caution is recommended in considering predictive testing for a disorder without curative, let alone meaningful, palliative treatment. Although for certain dominant disorders some 50 percent of individuals at risk may receive good news, the other 50 percent face, effectively, a death sentence. Given the remarkable pace of advances in human genetics, it may well be possible in the foreseeable future to develop a therapy that enhances the extant biologic mechanism already in place that delays the manifestations of later onset disease for decades after birth. No life should be ruined by severe depression or suicide only to discover later that a critical palliative remedy has emerged. Clearly, there are extraordinarily difficult circumstances related to planned childbearing in the face of 50 percent risks for a neurodegenerative disorder coupled with a wish not to know. In these special circumstances, predictive testing can be regarded as acceptable only if performed with extreme care, concern, and professionalism. Preconception care should begin during visits to the family physician after menarche. Early adolescence is also a vital period during which to inculcate the importance of genes and the wisdom of assimilating and updating information on family history. Linkage of family history to the common experience of physical and mental handicap, outlined in the context of personal risk in childbearing, provides a compelling and cogent framework on which physicians, teachers and parents can build. This preparatory background may help educate all women about the importance of planning pregnancy. Over 50 percent of pregnancies in the United States are not planned and are often unintended. The discovery or realization of nonpaternity at the time of prenatal diagnosis is fraught with potentially serious personal, medical, social, and legal problems. In the effort to do no harm, we have requested a counseling session with the prospective mother alone. If, however, testing of the misattributed partner has genetic implications, nondisclosure becomes legally untenable. Duty to warn Physicians and counselors traditionally owe no duty to individuals with whom they have never met or entered into any treatment relationship. Regents of the University of California),192 it has become clear that when a serious risk to the health or life of a third party is recognized, a duty of reasonable care evolves that demands protective action. For colorectal cancer there is evidence that over 50 percent of families at risk do not receive the necessary information. The loss of chance legal doctrine makes it incumbent upon geneticists/counselors to impress on their patients the need to warn blood relatives if a serious genetic threat is determined. Litigated examples include failure to warn of the risk of medullary thyroid cancer, familial adenomatous polyposis with colon cancer and the fragile X syndrome. Sudden death as a consequence of a monogenic disorder invokes specific responsibilities not only by the pathologist performing the autopsy but also the geneticist or genetic counselor, if involved with the family. Determination of the cause of sudden death, if not clearly obvious, may be ascribed to an arrhythmia. Where cardiac pathology points to a cardiomyopathy, similar considerations pertain. Counseling of next of kin in such cases is important, more especially since they may face a 50 percent personal risk. On occasion, a patient at high risk may refuse to be informed about a specific genetic test result. However, if that result implicates a specific disorder that not only places that individual at risk but as a consequence may cause harm to others, the ethical 20 Genetic Disorders and the Fetus imperative would demand communication of that unwanted information. Indications for preconception genetic counseling the indications for preconception genetic counseling should be determined at the first visit and can be considered in a few clear categories. Preconception genetic counseling It is an anachronism that preconception genetic counseling in the 21st century, despite being recognized as important, is not widely practiced. Physical examination and necessary special tests also focus on acquired and genetic disorders that could, during pregnancy, threaten maternal and/or fetal welfare. Previously undiagnosed/undetected disorders may be determined for the first time at this visit and may be important for planned childbearing and the selection of future prenatal diagnostic tests. There is a need to insist that the male partner attend the preconception visit (or absolutely the first prenatal visit), providing an opportunity to detect at least obvious genetic disorders and solidify information possibly provided earlier about his family history. In some countries, largely for economic reasons, older ages have been used as an indication for prenatal study. The maternal age guideline, while still an important marker for increased risk communication, is no longer sacrosanct. The advent of noninvasive prenatal screening has further assisted in enabling younger women to benefit from early prenatal diagnosis (see Chapter 11). Excluding infants with chromosome abnormalities, a prospective analysis of 102,728 pregnancies (including abortions, stillbirths and livebirths) in Texas found that the incidence of congenital malformations increased significantly and progressively in women after 25 years of age. A previous fetus or child with a genetic disorder A genetic evaluation and counseling are usually indicated when a previous fetus or child has or had a genetic disorder, unless the matter is straightforward. Failure or delay in the diagnosis of a monogenic disorder leaves the parents without the option of prenatal diagnosis in a subsequent pregnancy. In addition, it deprives them of the option of preimplantation diagnosis for those disorders with known mutations. Failure to make an early diagnosis of a genetic disorder during the first 5 years of life is common. For example, the Rotterdam Clinical Genetics Group reported that 50 percent of children affected by neurofibromatosis had been treated for related symptoms before a specific diagnosis had been made. Frequently, distressed parents will select a different physician for a subsequent pregnancy and a new or more recent insight may shed light on the cause of the previous disorder. Confined placental mosaicism may also be associated with intrauterine growth restriction,276 requiring serial ultrasounds during the pregnancy. Given the heterogeneous nature of genetic disease, being alert to alternative mechanisms of causation will on occasion be rewarding. Although nonpaternity is more likely, a judicious approach would also include consideration of uniparental disomy. The establishment of the molecular basis of recognized syndromes, previously undetectable prenatally, now provides new opportunities for couples seeking prenatal diagnosis. Examples abound and include some of the craniosynostosis syndromes, certain skeletal dysplasias and many other disorders. In one of our cases, a father with metaphyseal dysplasia of Schmid, troubled by the indignities and hurts of growing up with severe short stature, elected prenatal diagnosis at a preconception visit. Subsequent mutation analysis of conceived twins yielded a normal prenatal diagnosis result confirmed postnatally. The longestablished prenatal diagnosis for both presymptomatic and symptomatic neurodegenerative disorders continue to be expanded to include disorders such as amyotrophic lateral sclerosis and even frontotemporal dementia, by analysis of the C9orf72 22 Genetic Disorders and the Fetus gene. For example, a parent with tuberous sclerosis and normal intelligence could not be certain that an affected child would not have intellectual disability. This was especially evident in our series of 50 couples having prenatal diagnosis for tuberous sclerosis. Certain genetic disorders may (i) threaten maternal health in pregnancy, (ii) threaten fetal health and survival, or (iii) be aggravated by pregnancy. Genetic disorders that threaten maternal health Advances in medical care have resulted in more women affected by genetic disorders surviving to childbearing age and becoming pregnant. There are several genetic disorders affecting the mother that can be aggravated and worsened during pregnancy. Awareness of these disorders facilitates better preconception anticipatory guidance and expectant management during pregnancy. Metabolic disorders that may worsen include ornithine transcarbamylase deficiency, homocystinuria, acute intermittent porphyria, and lysinuric protein intolerance. Hyperammonemia during pregnancy/delivery or postpartum coma may be the presenting signs of a female heterozygote with ornithine transcarbamylase deficiency. In a study of 12 women with Loeys Dietz syndrome with 21 pregnancies, six had one of these major complications. Sophisticated and multidisciplinary care and counseling are necessary for women with Marfan syndrome. Pregnancy care: r All appropriate information that would have been reviewed in the preconception period should be discussed if counseling was shortly after pregnancy was established. Labor and delivery: r Planned delivery should occur in hospitals with available cardiac surgery and neonatal intensive care unit facilities. Caution should be exercised in using epidural anesthesia because of the often associated dural ectasia and/or the presence of scoliosis. Postpartum: r Women should be advised about the continuing risk of aortic dissection in the postpartum period with attention to all matters covered in previous counseling. Current recommendations suggest greatest therapeutic efficacy by combining a beta blocker with losartan to reduce the rate of aortic root dilatation. Women with short stature and Marfan syndrome appear to have an increased risk of aortic dissection and hence elective surgery would need earlier consideration. The first degree relatives of an affected woman with a thoracic aortic aneurysm have up to 30 percent likelihood of having or developing an aneurysm. Hypertension may be a problem for the pregnant patient with autosomal dominant polycystic kidney disease. Carriers of hemophilia A are best cared for by a high-risk perinatal obstetric group. Prenatal sex determination (whether or not prenatal diagnosis by mutation analysis is chosen) is important for the management of labor and delivery, with special reference to the possible need for cesarean section. In addition, vacuum-assisted delivery with an affected male could result in a massive cephalohematoma requiring blood transfusion.
References
- Sacco RL, DeRosa JT, Haley EC Jr, et al. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA 2001;285(13): 1719-28.
- Cioffi U, Bonavina L, De Simone M, et al: Presentation and surgical management of bronchogenic and esophageal duplication cysts in adults. Chest 113:1492, 1998.
- Stamm WE, Webb DI. Partial deficiency of hepatic glucose-6-phosphatase in an adult patient. Arch Intern Med 1975;135:1107.
- Berry M, Tzeng Y, Masland C: Percutaneous transtracheal ventilation in an obstructed airway model in post-apnoeic sheep. Br J Anaesth 113: 1039, 2014.