

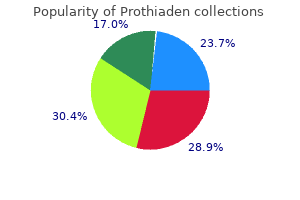
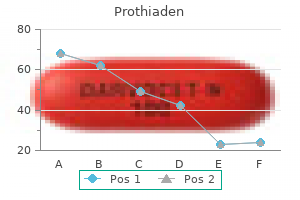
Prothiaden
| Contato
Página Inicial
Mihir K. Bhayani, MD
- Fellow, Department of Head and Neck Surgery
- University of Texas MD Anderson Cancer Center
- Houston, Texas
It most commonly consists of a few scattered symptoms zoloft overdose 75mg prothiaden purchase with amex, erythematous-to-brown plaques that are usually larger than 6 cm treatment coordinator 75mg prothiaden order mastercard. Histologic examination shows a superficial lymphocytic infiltrate with minimal nuclear atypia treatment definition best purchase prothiaden. Epidermotropism is scant or absent treatment math definition buy prothiaden 75 mg without prescription, and dermal fibrosis correlates with the chronicity of the process treatment without admission is known as prothiaden 75mg lowest price. Approximately 10%30% of patients ultimately develop an overt malignant transformation. There is a predilection for the head and neck area, especially the forehead, which has the highest density of pilosebaceous units. Histopathologic evaluation reveals cells in sebaceous glands often associated with destruction of hair follicle structures due to infiltration by a T-lymphocytic process. Even idiopathic cases Cutaneous Lymphoid Dyscrasias (Clonal Dermatitis) Cutaneous lymphoid dyscrasias or clonal dermatitis, which include a variety of lymphocyte-rich dermatoses, are often characterized by clonal T-lymphocyte proliferations that occasionally progress to bona fide mycosis fungoides. Clinically they exhibit a myriad of cutaneous presentations from poikiloderma (atrophic patches) to hyperpigmented areas resembling pigmented purpuric dermatosis or an acneiform presentation of follicular mucinosis. The hyperconvoluted nature of the nuclei is evident as complex nuclear folds seen through the chromatin. It typically manifests with a solitary, hyperkeratotic, often verrucous plaque on the lower limb. Biopsy results show atypical cerebriform lymphocytes with a perinuclear halo almost exclusively localized within the intraepidermal compartment. Although most cases have an indolent protracted course, generalized and sometimes aggressive variants have been reported. Cases presenting with a solitary lesion are extremely indolent and could be considered a reactive or pseudolymphomatous process. Low power (A) shows a moderately dense dermal infiltrate of lymphoid cells admixed with inflammatory cells, including neutrophils and eosinophils (B). The presence of Pautrier microabscesses, defined as four or more atypical lymphocytes arranged in an aggregate in the epidermis, is classic but is seen in only a minority of cases. The malignant T-cell clone often evolves into large-cell morphology during tumor progression, although rare cases show large-cell morphology from the early patch lesions. The presence of large cells in a skin lesion should be distinguished from "large-cell transformation," which displays rapid skin, node, and visceral progression, is refractory to treatment, and is associated with poor survival. Progression is associated with small-to-large clusters of atypical cells with preserved nodal architecture, followed by partial or total effacement of the node by neoplastic cells. Variable peripheral blood involvement can be demonstrated in all stages of skin disease, although it is most prevalent in patients with tumor or erythrodermic presentations. A staging bone marrow aspirate and biopsy is not recommended, unless unexplained cytopenias are seen. In an investigational setting, electron microscopy, cytogenetics, and molecular analyses have shown that a higher percentage of patients have occult involvement of internal organs. Plaque stage (A, C, D) demonstrates a band-like infiltrate (A, C) with some epidermotropism in the form of Pautrier microabscesses (D). In the tumor stage (B, E, F) there is a deep and dense infiltrate without significant epidermotropism. Borderline lesions are smaller but also tend to have a prolonged course, often with spontaneous resolution. The tumor has a favorable prognosis, and often complete or partial spontaneous regression occurs. In most instances, the tumor cells have anaplastic morphologic characteristics, showing round, oval, or irregularly shaped nuclei; prominent (eosinophilic) nucleoli; and abundant cytoplasm. Less commonly, the neoplastic cells have a pleomorphic or immunoblastic appearance. Reactive lymphocytes are often present, but infiltrating eosinophils are often less conspicuous. Radiotherapy is the preferred treatment for solitary or localized disease, with combination chemotherapy reserved for patients with generalized skin lesions or extracutaneous dissemination. Lymphomatoid Papulosis LyP is characterized by recurrent crops of self-healing, red-brown, centrally necrotic, asymptomatic papules and nodules. Sheets of tumor cells are present in the dermis (A) and are associated with marked pseudoepitheliomatous hyperplasia. The cells are quite varied and bizarre (B), and frequently show abnormal "embryoid" shapes (C) constituting the "hallmark" cells. Chapter85 T-CellLymphomas 1367 Histopathologic evaluation demonstrates that infiltrates are nonepidermotropic, with variable numbers of medium-sized to large pleomorphic T cells with or without cerebriform nuclei and immunoblasts. The infiltrate is often accompanied by a mixed infiltrate with reactive B cells and granulomatous or histiocytic component. Multiagent chemotherapy is used in most instances, with radiation therapy reserved for patients with localized disease. Patients typically present with a single red-purplish nodule or tumor involving the head and neck regions. In our opinion, cases presenting with a single lesion should not be diagnosed as having lymphoma; despite its clonal nature, the prognosis is invariably benign. These lesions were called pseudolymphomas in the past without significant consequences. Five-year survival rates are 100% in unilesional cases and exceed 60% in patients with more extensive disease. Sustained remissions have not resulted from radiation, immune therapy, or multiagent chemotherapy. Cytotoxic features such as necrosis, hemorrhage, and vasculitis are commonly encountered. Men are affected more commonly, and patients present with extensive erosive patches with frequent mucosal involvement. Occasionally the lesions are exophytic and hemorrhagic, resembling pyogenic granuloma. Histologically the infiltrate is markedly epidermotropic and adnexotropic, infiltrating into hair follicles and sweat glands, eventually becoming hemorrhagic and ulcerated. This condition is mostly reported in Asia and Latin America with rare cases seen in the United States. Although occasional cases remain localized in the skin, most of these lymphomas eventually involve other sites such as the testes or the gastrointestinal tract. A careful ear, nose, and throat evaluation to rule out nasopharyngeal involvement is important. The lesions are mostly large ulcerated tumor lesions with hemorrhagic and necrotic appearance. Histologically the tumor is composed of a deep infiltrate with intermediate-sized lymphocytes with cytotoxic changes, hemorrhage, and necrotic debris. A histologic landmark is the presence of angiocentric and angiodestructive features. A lymphoproliferative disorder resembling hydroa vacciniforme has been reported almost exclusively in Latin America, especially in the Andes region, where the combination of high altitude with intense ultraviolet rays triggers this process, which is mostly seen in vulnerable indigenous patients. These patients present with facial edema, hepatosplenomegaly, and necrotizing hemorrhagic lesions triggered by sun exposure or an arthropod bite reactions. These conditions are associated with a very aggressive clinical course and patients often succumb to hemophagocytic syndrome. Subcutaneous Panniculitis-Like T-Cell Lymphoma Subcutaneous panniculitis-like T-cell lymphoma is a rare entity. Patients, typically younger and female, present with asymptomatic deep subcutaneous nonulcerated nodules and plaques involving the legs. Overlapping or preceding signs of systemic lupus erythematosus or other autoimmune conditions are commonly observed. Histopathologic examination reveals a subcutaneous infiltrate with pleomorphic medium-sized T cells mixed with a reactive lymphoid infiltrate and some histiocytes. Differential diagnosis includes the frequently fatal but nonneoplastic cytophagic histiocytic panniculitis. The prognosis is fairly good, with a 5-year survival rate over 80% with the exception of patients with a concurrent hemophagocytic syndrome (fevers, cytopenias). Cases in which the panniculitic findings coexist with systemic lupus erythematosus may behave in a clinically indolent fashion. Primary Cutaneous -T-Cell Lymphoma Cutaneous T-cell lymphoma is a rare condition that tends to present with extensive panniculitis-like plaques on the extremities with a tendency to ulcerate during the course of the disease. Several of our patients had comorbidities associated with immune suppression, including autoimmune conditions or other lymphoproliferative conditions or malignancies. Chronic antigen stimulation has been hypothesized to play a role in the pathogenesis Lymphomatoid Granulomatosis Lymphomatoid granulomatosis is a rare multiorgan disease of the lungs, nasopharynx, joints, and peripheral and central nervous systems. Although nodules are most common, some patients have nonspecific macules, papules, or ulceration. Histologic evaluation reveals an angiocentric, polymorphous infiltrate of atypical lymphocytes and histiocytes surrounding and invading blood vessels within the dermis. However, reports have suggested a massive reactive T-cell infiltrate driven by a small number of clonal B cells. Although the clinical course is variable, the prognosis for patients with diffuse pulmonary involvement or evolution to high-grade lymphoma is poor, with a median survival of less than 2 years. Treatment depends on histologic findings and extent and location of disease, and may include corticosteroids, radiotherapy, and chemotherapy. Patients with disease that is not "acute" are considered to have "smoldering" disease and have lesions that may wax and wane in size and shape despite treatment. Despite some uncontrolled clinical trial results that have been reported to suggest "cures" in this disease, the general perception remains that this disease is not curable with standard therapies available today. The disease behaves similarly to other low-grade lymphomas, with periods of remission gradually becoming shorter with subsequent therapeutic interventions. A driving force in the development of treatments for this disease is the goal of altering the natural history for this group of poor-prognosis patients. No clinical trial has determined that aggressive early therapy is better than sequential palliative approaches or investigational approaches, and new treatments continue to be developed and tested for these patients. This classification was based on the evaluation of 347 patients and a multivariate analysis of potential prognostic factors. Investigators at the National Cancer Institute retrospectively analyzed 152 patients who underwent uniform pathologic staging. Good-risk patients had plaque-only skin disease without lymph node, blood, or visceral involvement, and a median survival of more than 12 years. Less than 10% of patients with stage 1A (limited patch) and less than 30% with stage 1B (extensive patch or plaque) progress to more advanced disease. Intermediate-risk patients had skin tumors, erythroderma, or plaque disease with lymph node or blood involvement (but no visceral disease) and a median survival of 5 years. Poor-risk patients had visceral disease or complete effacement of lymph nodes by lymphoma, and a median survival of 2. Flow cytometry allows the detection of cell populations with a normal (diploid) number of chromosomes versus abnormal (aneuploid) numbers, and cytogenetic analysis precisely identifies the individual chromosomal structure and number. Hyperdiploid cell clones were demonstrated in patients with large-cell histology, aggressive disease, and shortened survival time. Malignant lymphocytes often have convoluted or multilobed nuclei and can be detected in the peripheral blood in 75% of patients. T2 T3 T4 Node N0 N1 N2 N3 Patches, papules or plaques covering 10% of skin surface. Whether this conversion is secondary to prior therapeutic modalities used remains uncertain. Hence most adjuvant laboratory methods are not helpful at the precise time when they are most needed. No studies have demonstrated that one topical therapy is more effective than another, and patient and investigator preference remains the most important discriminating factor governing choice. Investigational approaches combining therapies also remains an active research strategy (see box on How We Manage Mycosis Fungoides/ Sézary Syndrome). The density of epidermal Langerhans cells in biopsy samples, as determined by immunoperoxidase stains, has been identified as a prognostic feature. Epidermal Langerhans cells are necessary for antigen recognition and processing in the normal immune response. Occasionally patients may develop a more clinically aggressive lymphoma concurrent with a change in the histologic appearance of the neoplastic cells and the pace of their disease. The mechanism of cell cytotoxicity for many cancer therapies involves the induction of apoptosis. Published guidelines include active drugs/treatment approaches, but unfortunately few randomized trials have been completed to allow for comparative effectiveness. Our philosophy regarding management is to try to avoid the side effects of therapy being worse than the symptoms from the disease. If these approaches do not control disease we favor attempting any of the systemic chemotherapy drugs or ideally an appropriate clinical trial. For younger/healthier patients with heavily pretreated refractory disease we consider earlier use of allogeneic stem cell therapy. However, macrophages appeared to be resistant to apoptosis induction and phagocytized apoptotic lymphocytes (but not nonapoptotic lymphocytes). Apoptosis induction may be the ultimate end point yielding benefit, but an immunologic effect due to monocyte phagocytosis and antigen presentation resulting from effector cells may also be present. The frequency of treatments can then be reduced, but some maintenance therapy (once every 24 weeks) may prolong the duration of remission. These studies all demonstrated high rates of remission in the early stage (patch or plaque stage of disease).
For patients with high levels of circulating disease treatment quotes generic prothiaden 75 mg overnight delivery, some studies may be performed with the use of peripheral blood samples; however symptoms exhaustion purchase on line prothiaden, assessment of a bone marrow biopsy and aspirate is essential world medicine 75mg prothiaden for sale. In addition chi infra treatment prothiaden 75 mg buy with visa, promonocytes in acute monocytic leukemia medications you cant take while breastfeeding buy prothiaden overnight delivery, megakaryoblasts in acute megakaryocytic leukemia, and abnormal promyelocytes in acute promyelocytic leukemia are added to the blast percentage. Flow cytometry utilizes multiparametric analysis of single cells to assess cellular granularity and size, cell surface, intracellular antigen expression, and other features. Flow cytometry can enumerate small populations of leukemic cells, below the limit of detection by morphology. This provides important prognostic data for risk stratification and informs therapeutic strategies. Cells from the aspirate are cultured, mitosis is interrupted, and the paired chromosomes are arranged to identify missing, translocated, or duplicated segments. In addition, use of targeted therapies is increasingly dependent on detection of a specific tumor genotype. With increasing numbers of genes to query, next-generation sequencing approaches offer advantages in sensitivity, cost, and efficiency over traditional testing methods. Large panels of genes can be tested simultaneously by pre-enriching for the targets of interest (by automated amplicon generation, or hybridization capture). With further improvements in analytical workflow and cost reduction, whole-genome and transcriptome sequencing could displace some existing diagnostic tools, as these platforms can provide simultaneous detection of mutations, gene expression, copy number alteration, and structural variation. These "preleukemic stem cells" may harbor the same class of mutations detected in individuals with clonally skewed hematopoiesis. The contribution of these preleukemic stem cells to chemotherapy resistance and relapse remains to be determined. Moreover, some mutations appear to be earlier, founding events, while other mutations are typically acquired during disease progression. The further identification of the sequence of mutation acquisition, as well as cooccurring and mutually exclusive genetic pathways, will help to better distinguish different subsets of this disease. Collectively, these lesions interfere with selfrenewal, differentiation, and survival pathways. Falini B, Mecucci C, Tiacci E, et al: Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. Pappaemmanuil E, Gerstung M, Bullinger L, et al: Genomic classification and prognosis in acute myeloid leukemia. The consequences of this malignant transformation include marrow failure, profound immune deficiencies, and not infrequently manifestations of a systemic inflammatory response of varying severity. Progress in basic, translational, and clinical research is cutting inroads into decade-old management paradigms. Several studies have suggested associations between leukemia diagnosis and season of the year. One study looking at this association found December and January as the two months with the highest number of new diagnoses, particularly for patients older than 65 years and men. Through the aggregate action of a network of mutated genes, these cells are characterized by impaired self-renewal properties, blocks in differentiation, limited capacity to undergo apoptosis, and altered signaling and metabolic pathways. Frequently, a given type of mutation is not sufficient to elicit the full leukemogenic phenotype and a second set of mutations is necessary for the fully transformed leukemic stem cells to develop. The transformation of immature hematopoietic cells that arises out of genetic changes in hematopoietic stem and more committed progenitor cells is thus considered a stepwise process in which genes of complementary mutation classes act together. In this model, two separate classes of mutations are operative: mutations that activate signal induction pathways and lead to uninhibited proliferation and survival; and mutations that affect transcription factors or other transcription elements that cause impaired hematopoietic differentiation and aberrant acquisition of self-renewal properties. Whereas mutations between these two groups easily coexist, mutations within one class are typically mutually exclusive. Although most of these more recently described genes do not neatly fit into one or the other mutation classes, the principle of synergistic activity in the process of malignant transformation remains nevertheless valid. This is important as older patients have more comorbidities, respond less well to chemotherapy than younger patients, and carry the worst prognosis of any age group. Blast percentage is best determined on a 500-cell differential of the marrow aspirate. Promyelocytes have moderately basophilic cytoplasm with numerous azurophilic granules, and monoblasts and promonocytes usually exhibit folded/convoluted nuclei and may contain prominent acidophilic nucleoli. Promyelocytes, promonocytes, and atypical pronormoblasts are considered blast equivalents in some subgroups. Cases with extensive fibrosis may represent a preceding myeloproliferative neoplasm or acute megakaryocytic leukemia. Patients typically present with a short history (18 weeks) of constitutional complaints (fatigue, lack of energy, malaise, profuse sweats), manifestations of bleeding (such as from gums, bruising in the skin, epistaxis, menorrhagia), or fevers. Fevers should always be presumed to be secondary to infections even in the absence of an identifiable focus and lead to rapid institution of antibiotic therapy. Extramedullary infiltrations of leukemia cells in the gingiva, skin, lymph nodes, or other organs occur occasionally. Signs at physical examination are often nonspecific but in their aggregate can lead to a correct diagnosis. Patients may demonstrate pallor, ecchymoses or petechiae, enlargement of lymph nodes, or rarely, hepatosplenomegaly. Leukocytosis of 100,000/µL or higher is considered an emergency and requires immediate efforts to reduce the disease burden. Abnormalities of renal and hepatic values may represent infiltration of these organs, even in the absence of clinical symptoms. Imaging studies are of little help in diagnosis but allow assessment of complications (pneumonia, cerebral bleed). The diagnostic workup consists of a morphologic assessment, immunophenotyping by flow cytometry, assessment of the karyotype, and a panel of gene mutations. Whereas morphologic assessment by itself is often not sufficient to render a diagnosis, flow cytometry will confirm the lineage assignment (myeloid vs. In the remainder, either no lineage-specific antigens are expressed (acute undifferentiated leukemia) or antigens of more than one lineage are present (mixed-phenotype acute leukemia). In the latter scenario, antigens of several lineages can be found on one (biphenotypic) or separate populations of blasts (bilineal). Karyotyping and gene mutation analysis may add diagnostic information in morphologically ambiguous situations but is otherwise of more interest in determining prognosis. The FrenchAmericanBritish classification described three types of blasts depending on the granule content (AC). A significant monocytic component is usually defined as 20% or greater of the nonerythroid elements and is usually required for making a diagnosis of acute myelomonocytic leukemia. The combination allows simultaneous evaluation of granulocytes (blue reaction product) and monocytes (orange-brown reaction product). A monocyte (top), a granulocyte (right), and a myelomonocytic hybrid cell that exhibits both the orange-brown and blue reaction products (bottom). They include blasts with long thin Auer rods (top left), immature cells with abnormal eosinophilic globules (top and bottom, second from left), abnormal salmon-colored granulation in the maturing cells, sometimes associated with a basophilic periphery (top and bottom, fourth from left), and slightly abnormal features in the mature neutrophils (far right). In fact, in some cases the blast count can be less than 20%, but the diagnosis of acute myeloid leukemia is still made with the cytogenetic finding of t(8;21). These granules are large, tend to cluster or coalesce, and are interspersed among the large eosinophilic granules, which are more difficult to see. As the eosinophils mature, the abnormal basophilic granules are less prominent and sometimes disappear (far right). A common misconception is that the basophilic granules are the granules of basophils and that the abnormal eosinophils are "hybrid cells. However, they are usually less prominent and less atypical than the basophilic granules of the abnormal eosinophils. Even within the same case, the granules can range from coarse, dark, and dense to fine and dust-like (B). The nuclei in the abnormal promyelocytes frequently exhibit a bilobed, dumbbell, or reniform shape. This is a diagnostically important feature, which can sometimes be difficult to recognize beneath the granules (B). Auer rods can be single, multiple, coalesced into Auer bodies, and even present in maturing cells (C). The microgranular type usually presents with an elevated white blood cell count (D). Although granules cannot be readily appreciated at the light microscope (E), granules can be demonstrated by electron microscopy. The abnormal nuclear shapes (bilobed, dumbbell, and reniform) can be easily appreciated (E). Bone core biopsy sample typically shows sheets of cells with abundant granular cytoplasm (F). These can be seen for weeks after therapy and do not signify a failed response to the differentiating agent. The blasts are megakaryoblasts (E) but in the bone marrow can have a sarcomatous appearance (F). The dysplastic forms have pale cytoplasm and abnormal nuclear shapes with hypolobation, hypersegmentation, and prominent nuclear excrescences. Dysplastic megakaryocytes can be recognized on the biopsy specimen by their abnormally small nuclei, which are sometimes multiple and widely spaced. Many patients receive complex therapeutic regimens, making this distinction difficult and impractical. In this case the touch imprints (A) and frozen section preparation (B) from a mass lesion in the cecal area from a 44-year-old patient were thought to represent a high-grade lymphoma. However, initial immunohistochemical stains did not support the diagnosis, and on closer inspection of the tumor and with subsequent immunomarkers, a diagnosis could be reached. A diagnostic clue to the origin was the presence of eosinophilic myelocytes (D), which indicate that some tumor cells have the capacity to differentiate into eosinophils. The former determine the likelihood of surviving induction chemotherapy and are strongly influenced by age, performance status, and a basic assessment of organ function (hepatic, renal, cardiac). Comorbidity indices (ideally as part of a more global geriatric assessment strategy) are particularly useful in older patients where the question of intensive chemotherapy versus lower intensity interventions assumes more significance. Whereas prognosis is hardly or not at all augmented by gene mutation testing in cytogenetically favorable and unfavorable groups, the situation is different in the intermediate-risk group, which is characterized by its size and heterogeneity in outcome. Ongoing efforts at whole-genome sequencing have identified additional repetitively mutated genes that are being considered in risk assessment with partly incongruous results. The blastic plasmacytoid dendritic cell tumor is an unusually aggressive malignancy that was previously called hematodermic malignancy. It frequently presents in the skin with a blastic proliferation of cells in the dermis (A and B), which inevitably spreads to the blood (C) and bone marrow (D and E). The European Leukemia Net has published a revised risk model based on integration of cytogenetic and gene mutation information (Table 59. Treatment of patients "unfit" for standard intensive induction therapy (often equaled to patients older than 60 to 65 years) is frequently approached differently from that of younger patients, and hence this chapter addresses therapy separately for each age group. In this population, characterization of the genotype has become helpful in guiding that decision. Leukemia has been at the forefront of the development of "targeted" therapy, mostly small-molecule drugs directed against defined intracellular proteins. InductionTherapy Patients Less Than 60 Years of Age Induction therapy is still built on the same two drugs as 40 years ago: cytarabine and anthracycline. Remission rates range from 60% to 80%, and long-term disease-free survival is about 35%. If the marrow continues to show blasts and is cellular, a reinduction is usually given. Remissions following reinductions are usually shorter lasting than remission after one induction cycle. The degree of neutrophil and platelet recovery at the time of remission has prognostic significance. Higher neutrophil and platelet counts at the time of remission are predictive of better relapse-free survival. In some cases a regenerating marrow may have an increased number of blasts, which may look like persistent leukemia. Further follow-up marrow studies will show reduction in blasts concomitant with a rise of neutrophils and platelets. Giving 10 instead of 7 days of cytarabine (3 + 10), increasing the dose of cytarabine from 100 mg/m2/day to 200 mg/m2/day, adding a third drug. A 43-year-old man presented with fatigue and was found to have a white blood cell count of 70,300/µL composed of mostly blasts. The patient was treated with standard induction chemotherapy, and a day-14 bone marrow study (DF) showed chemoablation effects with an empty marrow, stromal injury, dilated sinuses, and only scattered stromal cells and plasma cells with no obvious blasts. A bone marrow study performed at day 28 (GI) showed regenerative changes of trilineage hematopoiesis. Substitution of daunorubicin by doxorubicin produced more toxicity without added benefit. Idarubicin is a 4-demethoxy anthracycline analogue of daunorubicin, which results in increased lipophilicity and better cellular uptake compared with daunorubicin. A more recent follow-up of the study suggested that the benefit extends to all cytogenetic risk groups regardless of age. Daunorubicin 90 mg/m2 daily × 3 equals 270 mg/m2, which may prohibit further anthracyclines therapy (cumulative cardiotoxic dose of daunorubicin 360450 mg/m2). Likewise, higher dose compared with lower dose daunorubicin improved remission rates.
Humans become infected when they eat partially cooked or raw fish containing plerocercoids treatment meaning discount 75 mg prothiaden otc, which develop into adult worms in the jejunum in about 6 weeks symptoms anxiety discount prothiaden 75mg online, growing to a length of 10 m medicine zocor prothiaden 75 mg purchase with visa, with up to 4000 proglottids15; when these worms lay eggs symptoms lupus cheap 75mg prothiaden overnight delivery, the life cycle is repeated medications like abilify prothiaden 75mg purchase with mastercard. After ova have been identified in the stools, expulsion of the worms by praziquantel (10 to 20 mg/kg as a single dose taken orally) and cobalamin replenishment is curative. Patients suspected of having an inborn error of metabolism should be evaluated by specialized laboratories, such as the McGill University laboratory of Prof David Rosenblatt-a premier diagnostic center. This syndrome was first identified in patients with tetanus given nitrous oxide for up to 6 days. Megaloblastosis develops within 24 hours and lasts less than 1 week after a single exposure. Severe neurologic deficits have been reported after prolonged intraoperative exposure to nitrous oxide in patients with unsuspected cobalamin deficiency. Children present between 3 and 10 years of age with megaloblastic anemia and neurologic presentations with low serum cobalamin levels associated with mild, persistent, benign proteinuria (in 90% of cases). Because cubam also participates in the renal tubular absorption of albumin, this is the basis for proteinuria found in Imerslund-Gräsbeck syndrome. Although dependent on the population studied, the frequency of (silent) subclinical cobalamin deficiency in the United States is suspected to be 10 times higher than classic (overt) cobalamin deficiency that is found in 1% to 2% of the population. Nevertheless, the issue of subclinical cobalamin deficiency has been a vexing problem and a semantic dilemma. Many elderly persons may have various symptoms consistent with aging Chapter39 MegaloblasticAnemias 533 (including fatigue, cognitive changes, lower quality-of-life measures, and subtle symptoms of neuropathy) that cannot be directly attributed to cobalamin deficiency, despite the fact that these very symptoms are often seen in symptomatic cobalamin deficiency; often this triggers testing with a serum cobalamin test, and a borderline result that spontaneously reverts to normal, or minimally fluctuates above or below the cutoff value, or remains stable without change over many years generates a new set of problems, including the need to label this entity and thereby make clinical decisions. Although some experts do not feel obliged to treat, preferring to wait for overt symptoms, others feel ethically bound to treat even without overt clinical manifestations; indeed, such clinical manifestations can be very subtle and are detected only by sophisticated neurophysiologic or imaging studies that are expensive and impractical for routine clinical practice. In support of earlier therapy in this clinical setting, there is compelling clinical evidence that combined B-vitamin supplementation to reduce homocysteine can reduce brain atrophy177 and both cognitive and clinical decline178 (see section on Homocysteine and Mild Cognitive Impairment). After replenishing potentially depleted cobalamin stores with parenteral cobalamin therapy, oral supplementation for 4 to 6 months on and 4 to 6 months off may afford an adequate cobalamin status in most patients179 as an alternative to continuous therapy. A key factor is the cost of cobalamin (and the lack of side effects associated with cobalamin therapy); for example, parenteral cobalamin, which can be purchased on the Internet for $15 for each 10 mg/10 mL vial, would last a year after replenishing stores. The additional purchase of 30-gauge 1 2 -inch insulin U100 syringes for monthly subcutaneous injection could be less costly than even generic tablets of 1 mg taken daily. For example, in Benin, central Africa, the prevalence of folate deficiency anemia was 20%, and in Zimbabwe, 30% had low folate levels, whereas in Sudan it was nearly 60%. Even in the United States, before folate fortification of food, about 20% of the population had low-folate status, and in Venezuela, 30% had low-folate status before such fortification. Decreased availability of folate-rich foods (in winter, after natural disasters, or during the wet season in central Africa), poverty, various cultural or ethnic diets (consisting of maize, rice, or well-cooked beans and vegetables), and cooking techniques that destroy food folate, coupled with the anorexia that accompanies chronic illnesses, are just a few of the reasons for rapid development of folate deficiency. Folate fortification of foods in the West has led to widespread elimination of folate deficiency and related anemia,182,183 leading to questions of whether testing for folate deficiency is even justified. There is also increased urinary loss of folate in pregnancy (about 14 µg/day versus approximately 4. Poor preparation for pregnancy, with a poorly balanced diet and preexisting multifactorial nutritional anemia that remains unaddressed, is a major factor accounting for serious pregnancy complications and adverse birth outcomes. Low-folate status associated with short interpregnancy intervals or twin pregnancies also predisposes to preterm births. However, the vast majority (over 90%) of pregnant women in resource-poor countries consume less than the estimated average requirement of folate191; in addition, a substantial number also consume less than optimum amounts of several other minerals, such as iron, and micronutrients, including cobalamin, as noted earlier. For example, studies on women from groups with low socioeconomic status from North India29,30 have estimated that the daily intake of folic acid ranged between 75 µg and 167 µg, which is far lower than the 400 µg/day required to prevent birth defects. When given daily or even twice weekly, the combination of iron (100 mg elemental iron) and folic acid (0. This is all the more important because of results from experimental studies designed to define the influence of gestational folate deficiency on the fetus (discussed later). Thus pregnancy with poor folate intake is the most common cause of megaloblastic anemia in the world. It is instructive therefore to conceptualize cellular folate deficiency as arising from etiologic categories of decreased supply. However, in the same patient more than one mechanism may result in net folate deficiency. The precise contribution of one mechanism over the other is often not obvious, and specific tests to define each mechanism are not routinely available for clinical use. Megaloblastic manifestations of folate deficiency are discussed within the context of the history and physical examination (discussed later). Cases of neuropathy in adults attributed to folate deficiency are rarely encountered; when they are, the possibility of alcoholism with thiamine deficiency must be considered. In any case, every patient with neuropathy, myelopathy, or psychiatric manifestations associated with megaloblastosis must be investigated in detail to rule out cobalamin deficiency. Gastrointestinal megaloblastosis begets further folate malabsorption, which propagates a vicious cycle of folate deficiency in the short term and cobalamin deficiency in the long term. With the exception of drug-induced defects or inborn errors of folate metabolism that result in decreased use of intracellular folates, all causes, irrespective of mechanism, result in reduced net delivery of folates to normal proliferating cells. The incidence of folate deficiency varies from country to country and even within regions in the same country. Preferential delivery of folate to the fetus can cause or aggravate folate deficiency in the mother. Before the advent of routine folate supplementation during pregnancy, the incidence of megaloblastic marrows in the United States, Canada, and the United Kingdom during late pregnancy was about 25%, but in South India, it was about 55%. Multiparity (multiple frequent pregnancies with a prolonged state of negative folate balance) and hyperemesis gravidarum commonly lead to folate deficiency. Because the anemia of pregnancy is most frequently caused by iron deficiency, combined iron and folate deficiency (dimorphic anemia) is the more frequent clinical presentation. Increased use of folates by the newborn leads to a drop in serum folate levels by about 6 weeks of age. This drop is exaggerated in premature infants (who have feeding difficulties, infection, or hemolytic disease leading to pure folate deficiency); hence supplementation is routine for them. The fetal brain is dependent on sufficient provision of maternal folate during embryogenesis. Such studies57 indicate that folate deficiency will significantly compromise early pregnancy outcomes (including the rates of pregnancy, rate of implantation, and effects on the number of live births). Even lesser degrees of folate deficiency to only one-third of optimum dietary folate for 2 months before and throughout gestation in dams, which coincidentally mimics the extent of insufficient dietary folate availability among women in vast areas of Northern India,64 also resulted in subtle histologic aberrations and defects during murine fetal development. These data also suggest the existence of a sensitive window during fetal neurodevelopment when folate deficiency dysregulates the expression of certain genes and/or proteins, which leads to the imprinting of abnormal neural circuits in utero that predispose to anxiety in adulthood. In concordance with these studies in mice, a prospective cohort human study has reported that lower maternal folate status in early pregnancy was associated with childhood hyperactivity/inattention and peer problems in early childhood. Folate is also critical during the early postnatal period and deficiency (in rodents) results in apoptosis of cochlear cells leading to severe hearing loss196 and learning and memory deficits. For example, 18-month-old children of mothers who took folate supplements had less "internalizing" patterns of behavior (emotionally reactive, anxious/depressed, somatic complaints, withdrawn) and less "externalizing" syndromes involving attention problems and aggressive behavior compared to offspring of women who did not take folate supplements. The long-lasting benefit to the offspring of women who take iron and folic acid during the early stages of pregnancy appears to be a consistent theme; in Nepalese women, such supplementation provided significant benefits to the proper neurodevelopment of their babies in utero. Although this clinical study was unable to assign whether iron or folic acid was the more important, there is sufficient supporting experimental evidence in the literature for both being critically important. Indeed, these human parallels to murine studies are consistent with the Barker hypothesis on the developmental origins of disease. Finally, there is also evidence to suggest that suboptimal folate intake that leads to low-folate status during adolescence can affect cognition and academic achievement, independent of socioeconomic status. They arise from disturbances in neurulation that involve incomplete closure of neural tissues, leading to major midline defects. The neural tube, which begins as a tiny ribbon of tissue, normally folds inward to form a tube by the 28th day after conception. The expression of Chapter39 MegaloblasticAnemias 535 folate receptors on embryonic neural tube and neural crest cells as well as the critical bursts of proliferative activity and the need for folate to support cell proliferation have been discussed earlier. Thus it is critical for a woman to have enough folate in her body before conception (periconceptionally) to ensure sufficient availability for the embryo. Conversely, the use of folic acid antagonists (trimethoprim, triamterene, carbamazepine, phenytoin, phenobarbital, and primidone) during pregnancy increases the risk for these birth defects by twofold. This level was chosen to ensure that women of childbearing age would have an increase in folic acid intake of at least 100 µg a day, which is about 25% of the recommended daily intake. Subsequent evaluation has clearly demonstrated that fortification of food with folic acid has had multiple salutatory effects during human development and that major congenital abnormalities can be prevented. Because of an incomplete knowledge base among some women22,226 and their tendency to consume lowcarbohydrate foods (which are the very foods that are fortified), there is continued concern that this group is still not getting adequate amounts of dietary folate. This is the basis for recommendations to continue to educate women of childbearing age to take folic acid supplements at 400 µg/day (beyond what they are already receiving through folate fortification of food). Although regulations for mandatory fortification of wheat flour with folic acid are in place (in 53 countries by 2010), they have not been uniformly implemented. Anencephaly with complete rachischisis (top panel), open infected meningomyelocele (bottom left), and iniencephaly with cleft lip (bottom right). Molly Paul, Anatomy Department Museum, Christian Medical College and Brown Memorial Hospital, Ludhiana, Punjab, India. Affected patients can also present with a syndrome of reversible subacute combined immunodeficiency syndrome with hypogammaglobulinemia and recurrent infections. In the short term, malabsorption leads to folate deficiency, but later in the chronic (longer than 3 years) phase of the disease, cobalamin malabsorption contributes additional clinical manifestations of cobalamin neuropathy. Although less severe than in nontropical sprue, it is more extensive, involving the entire small intestine. After investigations for associated iron, cobalamin, and folate deficiencies, therapy with folate and a broad-spectrum antibiotic. However, the dramatic response to folate, which is curative in the first year in about 60% of cases (this cure is cited to be almost diagnostic of the disease), has not been explained. Moreover, folate-fortified foods do not reach all women of reproductive age adequately, particularly the most needy women in the lowest socioeconomic bracket. The recognition that folate deficiency developing in hemolytic disorders can lead to an acute aplastic crisis has led to recommendations favoring routine prophylactic administration of folate. A recent metaanalysis of 19 studies have put to rest the concern of iron-folate and malarial progression231 and a Cochrane analysis identified no evidence that this is a problem when combined with antimalarial drugs and insecticide-treated bed nets. An unexpected increase in transfusional requirement or a fall in platelets can also suggest folate deficiency. It results from a possibly inherited sensitivity to gluten (a Chapter39 MegaloblasticAnemias 537 glutamine-rich protein found in wheat, barley, rye, and other grains) and a related substance, gliadin. Patients present between the ages of 30 and 50 years with intermittent or persistent diarrhea (abrupt in 20%), weight loss, abdominal distention with discomfort, glossitis, and megaloblastic anemia. Diagnosis is established by documenting the presence of sensitive and specific serum antiendomysial antibodies IgA type or antitissue transglutaminase antibodies, malabsorption, and jejunal biopsy. These genetic polymorphisms merely reflect variants that are more frequent than the expected 1% allelic variation that could be found in any population. However, because disease association is not equivalent to disease causation, much more study is required to strengthen such relationships. A listing of the spectrum of these polymorphisms can be found in specialized texts. Acquired infantile-onset cerebral folate deficiency, which is associated with autoantibodies to folate receptor-,242,243 usually develops 4 to 6 months after birth and is characterized by agitation, insomnia, delayed development with deceleration of head growth, psychomotor retardation, cerebellar ataxia, pyramidal tract signs in the legs, dyskinesias (such as choreoathetosis and ballismus), a severe polyneuropathy, and in some cases, seizures. Untreated, central visual disturbances can become manifest and lead to optic atrophy and blindness by the third year. The folate receptor autoantibody titer decreases with restriction of bovine milk intake but promptly increases upon rechallenge. Deficiency-thiamine-responsive megaloblastic anemia (thiamine transporter 1 mutation) B. Alternatively, cobalamin deficiency may present with paranoid ideation, dementia, cognitive dysfunction, delusions, or lack of energy manifested by slowed responses. The family may indicate the progressive evolution of a marked personality change and may be able to help trace the evolution of symptoms and deviations from the time when the patient was last well. Although beer has higher folate content than other alcoholic beverages, alcoholism may lead to neglect of healthy dietary practices in favor of alcohol. Patients who have one nutritious meal each day tend to stave off the eventual development of folate deficiency. Alcohol consumption leads to a relatively rapid (2- to 4-day) fall in serum folate levels. Excess alcohol consumption is possibly the most common cause of folate deficiency in the United States. Methotrexate binds with high affinity to human dihydrofolate reductase and leads to trapping of folate as a metabolically inert form (dihydrofolate). Sulfasalazine produces megaloblastosis in up to two-thirds of patients taking full doses (over 2 g/day) by decreasing absorption of folates and induction of Heinz body hemolytic anemia. Whereas folates protect against spontaneous abortion,253 folic acid supplementation of women receiving antiepileptic drugs, which are known to interfere with folate absorption, also led to a significant reduction of spontaneous abortion. A recent randomized controlled trial among children 6 to 15 years of age who were initiated on phenytoin has provided incontrovertible evidence that taking folic acid 0. The only caveat is that before initiating long-term folic acid supplements, the cobalamin status must be normalized. Although antineoplastics and antiretroviral antinucleosides such as azidothymidine lead to megaloblastosis, the temporal sequence and investigations to rule out cobalamin or folate deficiency should easily lead to a correct causal assignment. Cardiopulmonary Gastrointestinal Dermatologic Genital Reproductive Psychiatric Neuropsychiatricc However, the neurologic spectrum of dysfunction in cobalamin deficiency is distinct. Inadequate hemoglobinization (from inadequate iron stores or globin synthesis) can mask the expected erythroid megaloblastic morphologic findings in the bone marrow and peripheral smear, and only specific therapy. Neuropsychiatric manifestations are not associated with megaloblastosis in up to 30% of patients.
They include a platelet count greater than 600 × 109/L after 3 months of at least 2 g/day of hydroxyurea (2 treatment 3 degree heart block order prothiaden overnight. In such situations medicine to increase appetite cheap prothiaden 75mg buy online, hydroxyurea can be substituted for (or combined with) other platelet-lowering agents 97140 treatment code generic 75 mg prothiaden amex. These criteria for hydroxyurea resistance are imperfect because they do not include the development of a thrombotic event while on therapy symptoms queasy stomach and headache prothiaden 75 mg purchase without a prescription, which is the central goal of therapy medicine 513 prothiaden 75mg purchase free shipping. Whether such patients who develop a new thrombosis would benefit from use of another therapeutic agent has not been explored. However, the leukemic risk increased significantly when the drug is used before or after treatment with alkylating agents, particularly busulfan. One can conclude from these studies that hydroxyurea therapy alone is less leukemogenic than alkylating agents or 32P alone, but a small increased risk for the development of leukemia secondary to its use cannot be completely excluded. These patients are reported to have a typical form of dysgranulopoiesis characterized by hypolobulated polymorphonuclear leukocytes with small vacuoles in neutrophils and p53 mutations. Chapter69 EssentialThrombocythemia 1119 An alternative to hydroxyurea is therapy with anagrelide. When studied in humans, it was noted that anagrelide in small doses produced thrombocytopenia. The drug acts primarily by reducing megakaryocyte size and ploidy, and decreasing megakaryocyte proliferation. Anagrelide therefore appears to lower platelet counts, primarily by interfering with the development of megakaryocytes. The effects of anagrelide do not involve thrombopoietin-mediated signal transduction events. Anagrelide in low doses is effective in lowering the platelet count in 93% of patients. Resistance to anagrelide therapy has not been documented, but occasionally patients have been observed to require extraordinarily high doses to control the excessive thrombocytosis. Excessive use will result in predictable thrombocytopenia and increase the likelihood of side effects. In addition, follow-up of more than 500 patients for more than 5 years had been reported. Anagrelide leads to a reduction in hematocrit in 2536% of patients, but it has no effect on white blood cell numbers, systemic symptoms, or the degree of splenomegaly. The most common side effects of anagrelide resulted from its vasodilatory and positive inotropic actions. These effects resulted in complaints of headache, dizziness, fluid retention, palpitations, and high-output cardiac failure. The vasodilatory effect leads to reduced renal blood flow, resulting in fluid retention. In addition, gastrointestinal complications, such as nausea, abdominal pain, and diarrhea, are prominent. These side effects usually develop within 2 weeks of initiation of therapy and frequently diminish in severity or resolve within 2 weeks of continued therapy. Because of its ability to promote fluid retention and the development of tachyarrhythmias, anagrelide therapy should be used with caution in patients with cardiac disease and should be administered carefully to elderly patients. If congestive heart failure or arrhythmias other than tachycardia develop, anagrelide therapy should be discontinued. Prolonged anagrelide therapy may be associated with a potentially irreversible drug-induced cardiomyopathy that is reminiscent of tachycardia-induced cardiomyopathy. Dose reduction can be used to lessen the degree of tachycardia or fluid retention. Before the institution of anagrelide, it is recommended that a basic cardiac evaluation be performed, including an assessment of cardiac risk factors, a careful cardiac history, and an electrocardiogram, which can be used as a baseline if the patient develops rhythm disturbances. Although most adverse effects are mild or moderate, in one series, therapy was discontinued in 16% of patients because of intolerable side effects, especially headache, nausea, fluid retention, and, rarely, frank congestive heart failure. Anagrelide has no mutagenic activity, but its use is not currently 100 Hydroxyurea + aspirin Event-free survival (%) 90 Anagrelide + aspirin 80 89% 84% 70 P =. Primary end points were arterial or venous thrombosis, serious hemorrhage, or death from any of these causes. Because of its small molecular weight, it is believed to be capable of crossing the placenta and thus may lead to fetal thrombocytopenia. Overall, patients randomized to anagrelide (and aspirin) were more likely to reach the composite primary endpoint of major thrombosis (arterial or venous), major hemorrhage, or death from vascular causes (p =. Anagrelide and aspirin seemed to offer at least partial protection from thrombosis because the prevalence of thrombotic events (8% at 2 years) was significantly less than that observed in the control arm of a previous study (28%). Intriguingly, the number of venous thromboses was less frequent in patients treated with anagrelide (p =. Also, the incidence of discontinuation of anagrelide therapy because of drug-related adverse events was significantly greater than that observed with patients receiving hydroxyurea. Therapy was administered to outpatients, most frequently at an initial dose of 3 million units daily, and usually produced a rapid decrease in platelets within 2 months. The mean time to complete response with a daily dose of 3 million units daily was about 3 months. In one study, 61% of patients required 3 million units three times a week, 15% once a week, and 24% daily. Side effects include flu-like symptoms during induction therapy, such as fever, bone and muscle pain, fatigue, lethargy, and depression. Normalization of blood counts occurred after a median time of 23 months; 12% of patients discontinued therapy because of inability to tolerate the drug, and 17% did not achieve normalization of their platelet counts. More importantly, no thromboembolic or hemorrhagic complications occurred during the period of treatment, although 12 thrombotic events occurred in 42 patients (24%) in the 24 months before the institution of therapy. Most patients were successfully treated with 4590 µg/weekly, and 22% of patients ceased therapy because of toxicity. Surprisingly, the hematologic and molecular responses persist for a number of months after discontinuation of therapy, suggesting that intermittent therapy may be an acceptable means of chronically administering this drug. The use of platelet antiaggregating agents remains an important area of investigation. In erythromelalgia, symptoms disappear for 24 days after administration of a single dose of aspirin. Although these agents surely have a role in the treatment of these specific complications, their use should be pursued with extreme caution because of the increased risk of hemorrhage. An independent analysis of the data from these two trials by the Cochrane Collaboration indicated that the use of low-dose aspirin therapy was associated with a statistically nonsignificant reduction in the risk of fatal thrombotic events and was not associated with an increased risk of bleeding episodes. Such patients who require aspirin benefit from the concurrent use of a proton pump inhibitor such as omeprazole rather than switching them to clopidogrel. Low-dose aspirin therapy must be restricted to patients with platelet counts of less than 1500 × 109/L and not be used in patients receiving anagrelide therapy unless they have experienced an arterial thrombotic event. The diagnosis of acquired von Willebrand syndrome should be excluded before aspirin use and considered a contraindication to the use of aspirin. If the clinician feels compelled to use some therapeutic intervention in young, asymptomatic patients, low-dose aspirin (81 mg/day) appears to be effective in the treatment of microvascular complications, and its use is associated with limited toxicity. Still, it seems reasonable to withhold therapy in younger, asymptomatic patients until the development of a clinically significant thrombotic or hemorrhagic event. Patients older than 60 years of age with other significant risk factors for cardiovascular complications are probably best served by immediate institution of therapy. In one large series, there was no significant relationship between the fetal outcome and the degree of maternal thrombocytosis or the presence of disease complications. In this series, there were no instances of excessive bleeding or other related complications during delivery. This group did not recommend the use of therapeutic platelet pheresis and, in fact, claimed that specific therapy (aspirin, heparin, or platelet pheresis) did not alter the clinical course. Low-dose aspirin (81 mg/ day), because of its profound effect on events involving the microcirculation such as erythromelalgia and transient ischemic events, has been used with increasing frequency in pregnant patients during the first and second trimesters. It is recommended that aspirin be discontinued at least 1 week before delivery to avoid bleeding complications such as an epidural hematoma during delivery or during the postpartum period. Because of the high risk of bleeding in patients with platelet counts greater than 1000 × 109/L with acquired von Willebrand syndrome, aspirin therapy is contraindicated. There is limited experience reported in the literature with aspirin therapy alone, and although the results are promising, the sample size is too small to confirm a beneficial effect. The observed true birth rate, however, was 75% in those receiving aspirin compared with 43% in the group in the literature who received no therapy. Chemotherapeutic drugs should be avoided during the period of conception and especially during the first trimester. Because the greatest risk of thrombosis is postpartum, thrombosis prophylaxis should be initiated in the form of low-molecularweight heparin and low-dose aspirin after delivery, unless the patient is hemorrhaging for approximately 8 weeks. Anagrelide therapy should be avoided in pregnant patients because of its potential to lead to fetal thrombocytopenia. Each of these agents in the normal population is associated with an increased incidence of arterial and venous thrombosis. Gangat et al retrospectively reviewed the consequences of such hormonal interventions in 305 women. Oral contraceptive therapy was associated with a high incidence of venous thrombosis occurring within the abdominal cavity, but estrogen-replacement hormone therapy in menopausal women did not appear to be associated with an increased incidence of thrombosis. This observation is surprising and might be a consequence of the limited numbers of patients included within this study. The thrombotic episode places a patient into a high-risk group in which myelosuppressive therapy is clearly indicated. Whether patients should have life-long anticoagulation or should receive only 612 months of anticoagulation after they are in a complete hematologic remission has not been investigated in a systematic fashion. Because the data necessary to make this decision are not available, this decision can only be made at the discretion of the treating physician. Antiplatelet therapy has been shown to reduce the risks of deep venous thrombosis and pulmonary embolism in a variety of high-risk groups. In patients who have a new thrombosis while they are in complete hematologic remission and are optimally anticoagulated, some consideration should be given to adding lowdose aspirin if the risk of hemorrhage is not excessive. Lastly, consensus response criteria have been updated recently to allow for more uniform reporting of therapeutic response within clinical trials (Table 69. Better means of identifying patients at risk for developing fatal thrombotic or hemorrhagic complications are necessary to provide the basis with which to develop the optimal care of such patients. The ability to reduce the incidence of thrombohemorrhagic episodes with cytoreductive therapy in highrisk patients is well established. The use of low-dose aspirin therapy to reduce the number of episodes of erythromelalgia and transient ischemic attacks is widely practiced, but whether aspirin therapy should be indiscriminately used remains a subject of dispute that will only be resolved with the completion of appropriately powered clinical trials. The rationale for this prospective clinical trial was based on laboratory findings of selective inhibition of malignant megakaryopoiesis compared with megakaryocytes isolated from normal controls after in vitro exposure to imetelstat. Grade 3 neutropenia was seen in 22% of treated patients, and lowgrade reversible transaminitis was seen in the majority of patients. The role of this infusional agent administered every 3 weeks in the Progressive disease Molecular response is not required for assignment as complete response or partial response. Partial response applies only to patients with at least 20% mutant allele burden at baseline. This is critical because the degree of reduction of platelet numbers has not served as a suitable biomarker for the development of these complications (see box on Personal Approach to Therapy of Essential Thrombocythemia). Therapy is geared toward interventions to reduce the potential for developing thrombotic episodes. Patients with the greatest risk of developing a thrombus have a number of characteristics, including age 60 years or older, history of a thrombotic event, leukocytosis (platelet count 11,000 × 109/L), and cardiovascular risk factors (hypertension, hypercholesterolemia, diabetes mellitus, obesity). Indiscriminant use of high doses of nonsteroidal antiinflammatory drugs should be avoided because this practice can lead to an increased risk of hemorrhage. In patients with a life-threatening thrombotic or hemorrhagic episode, plateletpheresis should be initiated in addition to starting them on hydroxyurea therapy. In high-risk patients, cytoreductive therapy has been shown to lessen the chance of developing additional thrombotic events with the reduction of extreme thrombocytosis to platelet counts below 600,000 × 109/L. High-risk patients include patients older than 60 years of age and patients with a history of a previous thrombotic episode, including erythromelalgia, transient ischemic attacks, or large-vessel thrombosis. Even though this treatment philosophy has been considered common practice, the recommendation to treat patients older than the age of 60 years who have not experienced a thrombotic episode with cytoreductive therapy is not based on robust data from multiple randomized trials. Asymptomatic high-risk patients without cardiovascular risk factors may not necessarily benefit from this treatment, and the decision on how to treat them should be based on individual assessment. At present, no therapy is indicated in asymptomatic patients younger than 60 years of age. If a patient has a platelet count greater than or equal to 1500 × 109/L and acquired von Willebrand syndrome with bleeding symptoms, platelet-reduction therapy is indicated to avoid the high risk of hemorrhage. In totally asymptomatic patients with platelet counts greater than 1500 × 109/L, we frequently observe the patients and do not feel compelled to treat them. Patients with acquired von Willebrand syndrome should clearly avoid the use of aspirin. Although we remain concerned about the leukemogenic potential of hydroxyurea, the risk appears to be low if not associated with the prior use of an alkylating agent. The development of malleolar ulcers is a frequent complication of hydroxyurea treatment and is a signal for the elimination of hydroxyurea as a therapeutic agent for that particular patient. Patients who initially receive hydroxyurea and no longer respond to this agent or experience toxicity and require another agent should not receive an alkylating agent. This sequence of administration is associated with an extremely high risk of leukemic transformation. Doses of each of these agents required for disease control will, of course, be dependent on the target platelet level that one hopes to achieve. Strict control to a platelet count of lower than 600 × 109/L does not appear to be necessary. In these patients, the addition of low-dose aspirin (81 mg/day) should be 1123 considered; it is less clear to us if patients who achieve better platelet control with cytoreductive therapy should also be so treated.
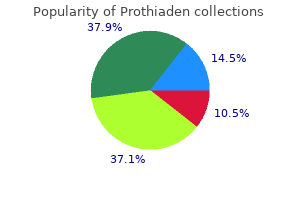
The main toxicity was dose-dependent thrombocytopenia symptoms glaucoma cheap 75 mg prothiaden, which occurred early in the treatment cycle (days 25 of a 21-day cycle) 3 medications that cannot be crushed discount prothiaden 75 mg mastercard. Venetoclax (Abt-199) was subsequently developed to replace navitoclax as a highly selective medicine effexor cheap prothiaden 75 mg with visa, orally available small-molecule Bcl-2 family protein inhibitor that binds with high affinity (Ki <0 treatment kitty colds generic 75mg prothiaden fast delivery. Once freed medications post mi cheap 75mg prothiaden with visa, E2F triggers the transcription of diverse genes involved in cell cycle progression (thymidylate synthase and dihydrofolate reductase, among numerous others). Regulation of the cell cycle has been found to be closely related to apoptosis, and disruption of normal cell cycle transit has been shown to be a potent cell death stimulus. A corollary of this observation is that agents that interfere with the cell cycle, in addition to blocking cell cycle progression, can be potent inducers of programmed cell death. Flavopiridol Flavopiridol (L86-8275) is a semisynthetic flavonoid derived from the Indian plant rohitukine. In addition to blocking cell cycle progression, flavopiridol has been shown to be a potent inducer of apoptosis in malignant hematopoietic cells. In clinical studies, flavopiridol was initially administered as a 72-hour continuous infusion every 2 weeks, with a maximally tolerated dose of 40 mg/m2. However, the occurrence of thromboembolic phenomena and the general lack of single-agent activity have limited enthusiasm for administering flavopiridol by this schedule. A novel pharmacologically directed schedule of flavopiridol has been developed in which half of the dose. Efforts are now underway to use this novel flavopiridol schedule in combination with other agents and in other hematologic malignancies. Because of limited single-agent activity, combination regimens involving flavopiridol are being explored in hematologic malignancies. More recently, evidence of synergism between flavopiridol and other signal transduction modulators has become the focus of considerable attention. Dose levels of 459 mg/m2 have proven to be tolerable; the maximum tolerated dose for either of these schedules has not been reached. Toxicities have been mild and include rash, nausea and vomiting, diarrhea, and fatigue. HypomethylatingAgents Promoter methylation within CpG islands regulates gene expression in all cells. Disruption of normal gene expression profiles accompanies malignant transformation, giving rise to complex patterns of gene expression. As a consequence, promoters of many genes have an altered pattern of methylation, resulting in either gene activation or gene repression. Azacitidine has been given after allogeneic stem cell transplantation, at lower doses, as a "maintenance" regimen, to prevent relapse. An oral formulation of azacitidine is currently in clinical development which would permit convenient and prolonged exposure to the drug, which may translate into better outcomes. Without the ability to fine tune histone binding to chromatin regions, gene expression is perturbed. Inhibition of this process by a group of agents termed histone deacetylation inhibitors alters gene expression in both normal and malignant cells. Each core histone has an N-terminal tail, which is lysine rich and positively charged. Specific amino acid residues at the N-terminal undergo a variety of enzymatic posttranslational modifications. Histone code is the name given to the combination of biochemical modifications affecting different histone residues that specify chromatin function. However, it has been suggested that various postsynthesis histone modifications be considered an epigenomic alphabet. Each modification is a letter, and the combination of modifications at a specified genomic region is a word that may have different functional meanings depending on the context. This directs these client proteins to polyubiquitylation and proteasomal degradation, thus contributing to the lowering of the threshold for apoptosis in cancer cells. Recently, we utilized donor-matched normal and transformed cells treated with vorinostat to identify a tumor cell-selective, proapoptotic gene expression signature containing effectors of the intrinsic apoptotic pathway that conferred tumor cell-selective apoptosis mediated by vorinostat and romidepsin. It is tempting to speculate that the cancer epigenome is altered in such a way as to predispose to altered expression of apoptotic genes in response to the transformation process, but definitive mechanistic evidence remains to be obtained. Short-Chain Fatty Acid Histone Deacetylase Inhibitors Sodium butyrate, a well-studied member of this class of compounds, induces in vitro growth arrest and differentiation of human leukemia cells at millimolar concentrations. Its clinical development has been hampered by its short half-life and difficulty in achieving millimolar levels in vivo. Phenylbutyrate, another derivative of butyric acid, is able to induce in vitro growth arrest and differentiation of leukemia cells at clinically achievable submillimolar concentrations. Importantly, at these levels, phenylbutyrate is able to synergize with retinoids in inducing cell cycle arrest, differentiation, and apoptosis of myeloid leukemia cells, and with ara-C in myeloid leukemias. No responses were noted in either study, with neurotoxicity being the dose-limiting toxicity. The objective response rate was 30% based on a standardized scoring system, and the median time to tumor progression was 202 days. In these studies, the common toxicities were diarrhea (52%), fatigue (52%), nausea (41%), and anorexia (24%). Although the definitive mechanism of action of this high degree of response is not known, a recent review outlines the current hypotheses. Furthermore, highly significant synergy has been observed in preclinical models combining vorinostat with bortezomib. A number of such studies are underway, but none to date has generated significant results showing combined agent clinical synergy. Exposure to panobinostat is associated with hyperacetylation of H3, H4, and Hsp90; increase in p21; and induction of cell cycle G1 phase accumulation. Other toxicities included nausea, diarrhea, vomiting, hypokalemia, and thrombocytopenia. Eight out of 11 patients with peripheral blasts had transient reductions in blast counts, which increased shortly after drug discontinuation. However, unlike vorinostat, single-agent activity has been seen, with response rates of almost 40% in refractory Hodgkin lymphoma. Even more impressive have been recent results in Hodgkin lymphoma patients relapsing after autologous transplantation, in which overall responses of greater than 70% and objective and durable responses were seen in 27% in a large cohort of 129 patents. A phase I trial tested entinostat in patients with refractory solid tumors and lymphomas. Prolonged disease stabilization was seen in some patients, and the drug was well tolerated and demonstrated antitumor activity. It is a natural product derived from Chromobacterium violaceum, a bacterium isolated from Japanese soil samples. Additionally, romidepsin was shown to exert antiangiogenic effects by modulating the expression of genes involved in angiogenesis. Cyclic Tetrapeptide Histone Deacetylase Inhibitors treatment permits a broad integration into currently approved chemotherapy regimens. Treatment-emergent adverse events that occurred in at least 20% of patients in the 0. Laboratory abnormalities reported included hypoalbuminemia and hepatic transaminitis. For example, when mocetinostat was tested in patients with relapsed Hodgkin lymphoma four patients died, of which two were treatment-related deaths. Protein translation is divided into three phases: initiation, elongation, and termination. Hematologic toxicity was frequent (neutropenia 44%, anemia 39%, and thrombocytopenia 76%), particularly in the initial stages of treatment, and was managed by reducing the number of days of drug administration. The most common nonhematologic toxicities were infections, diarrhea, nausea, and fever. Severe nonhematologic toxicities included infection (10%), fatigue (5%), and increased alanine aminotransferase (3%). They form multimolecular complexes with cellular proteins (called clients), regulating their correct folding, repair, degradation, and function. Newer, nongeldanamycin agents have improved bioavailability and water solubility, and have been observed to have limited single-agent activity against hematologic malignancies. The Goldie-Coldman hypothesis predicts that drug-resistant tumor cell clones survive because of a favorable spontaneous mutation, which occurs in approximately one in a million cells. Because 1 g of tumor contains 1 × 109 cells, it becomes obvious that high tumor burden states contain cells with a tremendous number of mutations, which can contribute to drug resistance. This is the rationale for using combination chemotherapy at specific dose intervals to maximize dose intensity. Drug-resistance mechanisms have been discovered and subsequently defined at the molecular level by investigators working in vitro with tumor cell lines selected in the presence of specific antitumor agents, by analysis of primary samples of untreated and treated hematologic malignancies, and through screening of tumor banks. Classes of resistance include acquired protein deficiency, loss of sensitivity to apoptotic signals, and age-related defects in the cellular pathways that normally lead to apoptosis. ProteinTranslationInhibitors the increased proliferation and cell survival of cancer cells imposes a requirement for increased levels of short-lived proteins involved in cell division and survival. This phosphorylated glycoprotein has a molecular mass of approximately 170 kDa and is localized to the plasma membrane, where it functions as a drug efflux pump (Table 57. To accomplish the former, this "hydrophobic vacuum cleaner" must detect drugs and expel them while they are still in the plasma membrane. A computer graphic representation of the three-dimensional reconstruction is shown as a shaded surface representation of the structure. Arrow indicates asymmetric opening providing access from the lipid phase to the aqueous core of the protein. This large aqueous chamber in the membrane that is open to the extracellular space is closed on the cytoplasmic side, presumably by the two 3-nM intracellular lobes (putative nucleotidebinding domains) and the hydrophilic cytoplasmic loops between the transmembrane domains. Thus, this large pore has a "gate" on the cytoplasmic side of the membrane that can regulate the transport of different-sized substrates. These tissues include the adrenal cortex, renal proximal tubule epithelium, biliary hepatocytes, small and large intestinal mucosa, pancreas, and endothelial cells of the brain and testis. The cell lines with the highest levels of drug resistance were found to overexpress all three proteins. This transporter has 18 transmembrane domains (12 in the amino end and six in the carboxyl end) and is coded on human chromosome 16p13. Low levels of resistance have also been reported for taxol, vinblastine, and colchicine. These agents appear to inhibit the function of this transporter, but whether this will translate into clinical impact has not received prospective attention. Transient hyperbilirubinemia was seen in 62% of the patients; these same patients had increased serum daunorubicin levels and a higher response rate. Although the response rate was not affected, the duration of remission was much shorter when these proteins were overexpressed. There is a striking correlation between drug resistance and alkyltransferase activity. Finally, cytokinetic factors represent a common theme in the case of most (but not all) antimetabolites, in that a reduction in the S-phase fraction generally leads to reduced drug sensitivity. Note that these resistance mechanisms are agent specific and are distinct from the more general modes of resistance. As discussed in the previous sections, these agents can inhibit with relative selectivity cellular signaling pathways essential for the survival of neoplastic cells. However, development of resistance has been observed in vitro and in clinical practice, and appears to be the rule rather than an exception with these agents. The mechanisms of resistance related to drug influx/efflux also can affect sensitivity to targeted agents. In addition, several other mechanisms have been described for signaling inhibitors, including genetic alterations, changes in protein expression, and activation of alternative pathways. Base Excision Repair Genetic Modifications Leading to Resistance to Signaling Inhibitors Cancer cells treated with kinase inhibitors tend to acquire genetic modifications that overcome the inhibitory effects of these agents. The development of resistance against a specific inhibitor can be the result of a preexisting cancer cell subpopulation carrying the mutation; once exposure to the drug occurs and sensitive cells die, this cell population experiences selective advantage. On the other hand, cell line studies have been able to induce the emergence of resistance to specific signaling inhibitors, suggesting that the genomic instability experienced by neoplastic cells facilitates emergence of new mutations that may affect drug sensitivity. Mutations that confer resistance to a kinase inhibitor commonly affect the affinity of the drug for the kinase domain without affecting its catalytic activity. Many signaling inhibitors target pathways that present amplification of one of its components. Further increases in gene amplification can affect the drugtarget balance in favor of the latter. Gene amplifications resulting in increases in target may be overcome by increasing the inhibitor dose, as it is done in clinical practice with imatinib; however, this is limited by the higher potential for adverse events. This can occur when mutations or amplifications result in increased activity or expression of signaling molecules located downstream or parallel to the point of inhibition. These "escape" mechanisms may be taken into consideration when designing combination strategies for treatment with multiple targeted agents. For example, many antimetabolites are prodrugs in that they must be converted intracellularly into active nucleotide forms to exert their cytotoxic actions. Consequently, events that interfere with cellular accumulation of drug or nucleotide formation will reduce activity. A third mechanism of resistance stems from the presence of increased intracellular levels of a competing metabolite. Fourth, alterations in the level of activity of a target enzyme or the presence of a mutant form that is a poor target of inhibition will also confer resistance. The continuing expansion of the therapeutic armamentarium with the addition of target-based therapies presents clinicians and researchers with several challenges.
Prothiaden 75 mg purchase with visa. HIV - AIDS - signs symptoms transmission causes pathology.
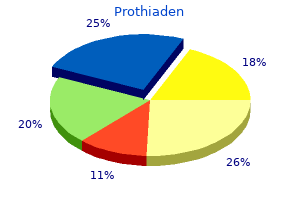
References
- Vassallo M, Shepherd RJ, Iqbal P, Feehally I. Age-related variations in presentation and outcome in Wegener's granulomatosis. J R Coll Physicians Lond 1997;31(4):396-400.
- Scheer B, Perel A, Pfeiffer UJ. Clinical review: complications and risk factors of peripheral arterial catheters used for haemodynamic monitoring in anaesthesia and intensive care medicine. Crit Care. 2002;6:199-204.
- Runge MS, Bode C, Matsueda GR, et al. Antibody-enhanced thrombolysis: Targeting of tissue plasminogen activator in vivo. Proc Natl Acad Sci U S A 1987;84:7659-62.
- Ahringer J. NuRD and SIN3 histone deacetylase complexes in development. Trends Genet 2000;16(8):351-356.
- Heidenreich A, Pfister D, Porres D: Cytoreductive radical prostatectomy in patients with prostate cancer and low volume skeletal metastases: results of a feasibility and case-control study, J Urol 193(3):832n838, 2015.